刘小泽写于19.5.7
主要看流程,这一篇不涉及真实数据展示
总的来说,Cell Ranger主要的流程有:拆分数据 mkfastq、细胞定量 count、定量组合 aggr、调参reanalyze,还有一些小工具比如mkref、mkgtf、upload、sitecheck、mat2csv、vdj、mkvdjref、testrun
首先是mkfastq 拆分数据
虽然这里用不到(因为我们下载的就是fastq数据),但是为了流程的完整还是要学习一下
目的:将每个flowcell 的Illumina sequencer's base call files (BCLs)转为fastq文件
特色: 它借鉴了Illumina出品的bcl2fastq,另外增加了:
- 将10X 样本index名称与四种寡核苷酸对应起来,比如A1孔是样本
SI-GA-A1,然后对应的寡核苷酸是GGTTTACT, CTAAACGG, TCGGCGTC, and AACCGTAA,那么程序就会去index文件中将存在这四种寡核苷酸的fastq组合到A1这个样本 - 提供质控结果,包括barcode 质量、总体测序质量如Q30、R1和R2的Q30碱基占比、测序reads数等
- 可以使用10X简化版的样本信息表
它的示意流程:

两种使用方式:
# 第一种
$ cellranger mkfastq --id=bcl \
--run=/path/to/bcl \
--samplesheet=samplesheet-1.2.0.csv
# 第二种
$ cellranger mkfastq --id=bcl \
--run=/path/to/bcl \
--csv=simple-1.2.0.csv
# 其中id指定输出目录的名称,run指的是下机的原始BCL文件目录
# 重要的就是测序lane、样本名称、index等信息
samplesheet.csv文件就是illumina常规使用的,类似下面这种。它除了需要指定各种ID、name之外,还要根据不同的试剂盒版本调整[Reads]长度
V2试剂盒R1序列长度为26bp(包括16bp的barcode+10bp的UMI),R2为98bp;
V3试剂盒R1序列长度为28bp(包括16bp的barcode+12bp的UMI),R2为91bp

还有一种10X定制的简单化的csv文件,例如:
Lane,Sample,Index
1,test_sample,SI-GA-A3
# 其中第一列指定lane ID,第二列是样本名称,第三列是index名称
使用简化版的这个文件,可以识别使用的试剂盒版本,然后自行调整reads的长度信息
最后的结果就是三个文件:I1序列文件以及两个测序文件R1、R2
目录结构如下:
- tiny-bcl/outs/fastq_path/bcl/
- Sample1
- Sample1_S1_L001_I1_001.fastq.gz
- Sample1_S1_L001_R1_001.fastq.gz
- Sample1_S1_L001_R2_001.fastq.gz
自己分析的数据也要改成这种结构存放,方便后续分析
小Tip--指定fastq文件位置
后续分析需要指定fastq位置,但是这些fastq文件可以由
cellranger mkfastq得到,也可以利用s Illumina'sbcl2fastq、公共数据、10X的bamtofastq,每种情况可能得到的fastq存放位置是不同的,那么如何根据不同情况进行指定呢?
第一种情况:
利用mkfastq或者bcl2fastq生成的文件,大概长这样

# 会有这几种选择方式[注意几种参数的设置]
# 1.所有mkfastq生成的样本
--fastqs=MKFASTQ_ID/outs/fastq_path
# 2. 多个flowcell生成的所有样本
--fastqs=MKFASTQ_ID/outs/fastq_path1,MKFASTQ_ID/outs/fastq_path2
# 3.所有bcl2fastq 生成的样本
--fastqs=/PATH/TO/bcl2fastq_output
# 4. 所有lanes上的test_sample1样本
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=test_sample1
# 5. lane1上的test_sample1样本
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=test_sample1 \
--lanes=1
# 6. 将test_sample1和test_sample2各自进行操作
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=test_sample1,test_sample2
其实从上面的各种设置也能看出来,一开始的样本命名规则是非常重要的
第二种情况:
也是利用mkfastq或者bcl2fastq生成的文件,但是同一个样本的数据放在不同的目录

# 1. 将所有SI-GA-A1样本的reads组合
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=SI-GA-A1_1,SI-GA-A1_2,SI-GA-A1_3,SI-GA-A1_4
# 2. 只处理SI-GA-A1样本的第一个index样本
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=SI-GA-A1_1
第三种情况:
也是利用mkfastq或者bcl2fastq生成的文件,但和Reports、Stats在同一个目录
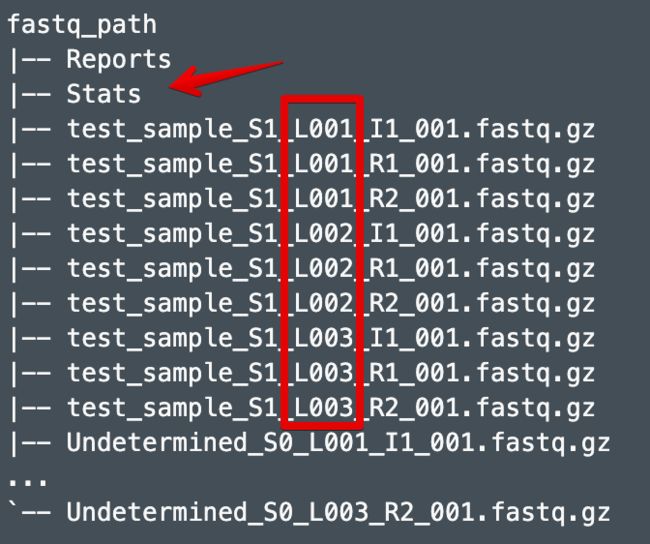
# 1. mkfastq得到的所有样本
--fastqs=MKFASTQ_ID/outs/fastq_path
# 2. bcl2fastq得到的所有样本
--fastqs=/PATH/TO/bcl2fastq_output
# 3. test_sample样本的所有lanes
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=test_sample
# 4. test_sample样本的lane1
--fastqs=MKFASTQ_ID/outs/fastq_path \
--sample=test_sample \
--lanes=1
第四种情况:
使用 mkfastq or bcl2fastq 得到的fastq文件和Report、Stats不在同一个目录,但命名方式与之前一样,这个目录中只能看到fastq文件
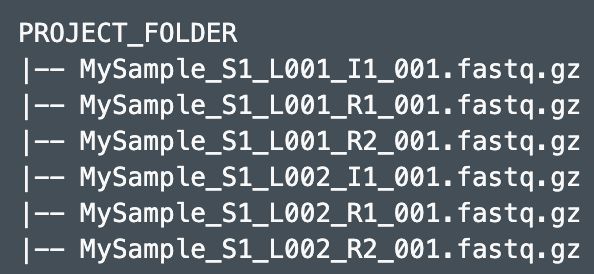
# 1.处理所有样本
--fastqs=/PATH/TO/PROJECT_FOLDER
# 2. 处理Mysample样本的所有lanes的数据
--fastqs=/PATH/TO/PROJECT_FOLDER \
--sample=MySample
# 3. 只处理Mysample样本的lane1数据
--fastqs=/PATH/TO/PROJECT_FOLDER \
--sample=MySample \
--lanes=1
第五种情况:
fastq命名方式变了,类似于这样:

它一般是从demux流程中拆分出来的数据,但是目前被mkfastq取代,没有好的方法,需要知道样本相关的index或者oligos
# 1.所有样本
--fastqs=/PATH/TO/PROJECT_FOLDER
# 2.所有SI-GA-A1样本
--fastqs=/PATH/TO/PROJECT_FOLDER \
--indices=SI-GA-A1
# 3.所有SI-GA-A1样本的lane1数据
--fastqs=/PATH/TO/PROJECT_FOLDER \
--indices=SI-GA-A1 \
--lanes=1
# 4.利用oligo
--fastqs=/PATH/TO/PROJECT_FOLDER \
--indices=AACCGTAA,CTAAACGG,GGTTTACT,TCGGCGTC
第六种情况:
数据命名与上面完全不同,因此需要自己重命名,方式就是
# 这个在单细胞实战(二)中介绍过
[Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz
# 其中Read Type
# I1: Sample index read (optional)
# R1: Read 1
# R2: Read 2
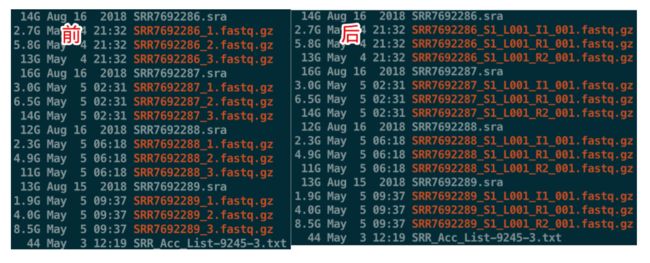
分析时就可以直接调用了
# 1.所有样本
--fastqs=/PATH/TO/PROJECT_FOLDER
# 2. 某个样本的所有lanes数据
--fastqs=/PATH/TO/PROJECT_FOLDER \
--sample=SAMPLENAME
# 3. 某个样本的某个lane
--sample=SAMPLENAME \
--fastqs=/PATH/TO/PROJECT_FOLDER \
--lanes=1
然后是count 细胞定量
这个过程是最重要的,它完成细胞与基因的定量,它将比对、质控、定量都包装了起来,内部流程很多,但使用很简单
先学会使用
每个版本要求的参数是不同的,尤其是V2与V3版本存在较大差异,这里先对V2进行了解
基本上自己需要输入的参数是:
# 这是示例,不是真实数据 #
cellranger count --id=sample345 \
--transcriptome=/opt/refdata-cellranger-GRCh38-1.2.0 \
--fastqs=/home/scRNA/runs/HAWT7ADXX/outs/fastq_path \
--sample=mysample \
--expect-cells=1000 \
--nosecondary
# id指定输出文件存放目录名
# transcriptome指定与CellRanger兼容的参考基因组
# fastqs指定mkfastq或者自定义的测序文件
# sample要和fastq文件的前缀中的sample保持一致,作为软件识别的标志
# expect-cells指定复现的细胞数量,这个要和实验设计结合起来
# nosecondary 只获得表达矩阵,不进行后续的降维、聚类和可视化分析(因为后期会自行用R包去做)
它的输出文件有很多
Outputs:
- Run summary HTML: /opt/sample345/outs/web_summary.html
- Run summary CSV: /opt/sample345/outs/metrics_summary.csv
- BAM: /opt/sample345/outs/possorted_genome_bam.bam
- BAM index: /opt/sample345/outs/possorted_genome_bam.bam.bai
- Filtered gene-barcode matrices MEX: /opt/sample345/outs/filtered_gene_bc_matrices
- Filtered gene-barcode matrices HDF5: /opt/sample345/outs/filtered_gene_bc_matrices_h5.h5
- Unfiltered gene-barcode matrices MEX: /opt/sample345/outs/raw_gene_bc_matrices
- Unfiltered gene-barcode matrices HDF5: /opt/sample345/outs/raw_gene_bc_matrices_h5.h5
- Secondary analysis output CSV: /opt/sample345/outs/analysis
- Per-molecule read information: /opt/sample345/outs/molecule_info.h5
- Loupe Cell Browser file: /opt/sample345/outs/cloupe.cloupe
Pipestance completed successfully!
从上到下依次来看:
- web_summary.html:官方说明 summary HTML file
- metrics_summary.csv:CSV格式数据摘要
- possorted_genome_bam.bam:比对文件
- possorted_genome_bam.bam.bai:索引文件
- filtered_gene_bc_matrices:是重要的一个目录,下面又包含了 barcodes.tsv.gz、features.tsv.gz、matrix.mtx.gz,是下游Seurat、Scater、Monocle等分析的输入文件
- filtered_feature_bc_matrix.h5:过滤掉的barcode信息HDF5 format
- raw_feature_bc_matrix:原始barcode信息
- raw_feature_bc_matrix.h5:原始barcode信息HDF5 format
- analysis:数据分析目录,下面又包含聚类clustering(有graph-based & k-means)、差异分析diffexp、主成分线性降维分析pca、非线性降维tsne
- molecule_info.h5:下面进行aggregate使用的文件
- cloupe.cloupe:官方可视化工具Loupe Cell Browser 输入文件
一些内置软件和算法
基因组比对—是否在外显子?
利用了 STAR比对工具,这款比对工具比对速度快,灵敏度高,是ENCODE、GATK推荐使用的工具,允许基因的可变剪切。比对完之后,利用GTF文件将reads溯源回外显子区、内含子区、基因间区:如果一条read的50%以上与外显子有交集,那么就认为它在外显区;如果不在外显子区,与内含子有交集,那么就认为它在内含子区;与外显子、内含子都没有交集,那么就认为在基因间区
MAPQ 辅助判断—在外显子的正确率有多少?
如果reads比对到了一个外显子区,同时也比对到了1个或多个的非外显子区,更相信它在外显子区,然后看MAPQ值,值越大越可信,如果MAPQ的值为255的话,那么就可以非常确定它比对到了外显子区
MAPQ即mapping quality,告诉我们这个read比对到参考基因组上某个位置的可信度,它的公式是:
-10logP(error),如果这个值大于30就认为比对发生错误的概率是千分之一
转录组比对—是否特异比对?
如果上面得到的外显子区域reads同时比对上有注释转录本上的外显子,并且在同一条链上,那么认为这个reads也比对到了转录组;如果只比对到单个基因的注释信息,那么认为它是特异比对到转录组的(uniquely /confidently mapped ),这样的reads才会拿来做接下来的UMI 计数
重点和难点在于自主构建参考信息
Cell Ranger为比对和定量提供了参考基因组及注释 pre-built human (hg19, GRCh38), mouse (mm10), and ercc92 reference packages
但是很多时候,我们需要根据自己的需要,自定义一套参考信息,但需要注意以下问题:
- 参考序列只能有很少的 overlapping gene annotations,因为reads比对到多个基因会导致流程检测的分子数更少(它只要uniquely mapped的结果)
- FASTA与GTF比对和STAR兼容,GTF文件的第三列(feature type)必须有exon,过滤后的GTF只包含有注释的基因类型
首先利用mkgtf过滤GTF文件
先从 ENSEMBL或UCSC上下载,然后使用mkgtf
cellranger mkgtf input.gtf output.gtf --attribute=key:allowable_value
# 其中键值对可以指定多个,比如
$ cellranger mkgtf Homo_sapiens.GRCh38.ensembl.gtf Homo_sapiens.GRCh38.ensembl.filtered.gtf \
--attribute=gene_biotype:protein_coding \
--attribute=gene_biotype:lincRNA \
--attribute=gene_biotype:antisense \
--attribute=gene_biotype:IG_LV_gene \
--attribute=gene_biotype:IG_V_gene \
--attribute=gene_biotype:IG_V_pseudogene \
--attribute=gene_biotype:IG_D_gene \
--attribute=gene_biotype:IG_J_gene \
--attribute=gene_biotype:IG_J_pseudogene \
--attribute=gene_biotype:IG_C_gene \
--attribute=gene_biotype:IG_C_pseudogene \
--attribute=gene_biotype:TR_V_gene \
--attribute=gene_biotype:TR_V_pseudogene \
--attribute=gene_biotype:TR_D_gene \
--attribute=gene_biotype:TR_J_gene \
--attribute=gene_biotype:TR_J_pseudogene \
--attribute=gene_biotype:TR_C_gene
# 这样得到的Homo_sapiens.GRCh38.ensembl.filtered.gtf结果中就不包含gene_biotype:pseudogene这部分
然后利用mkref构建参考索引
# 基本使用(单个物种)
cellranger mkref --genome=hg19 --fasta=hg19.fa --genes=hg19-filtered-ensembl.gtf
# 可以使用--nthreads使用多线程加速
# 得到的输出结果(保存在--genome名称的目录中)
ls hg19
fasta/ genes/ pickle/ reference.json star/
# 如果对于多个物种组合(本文的数据其实就应该这样组合起来)
cellranger mkref --genome=hg19 --fasta=hg19.fa --genes=hg19-filtered-ensembl.gtf \
--genome=mm10 --fasta=mm10.fa --genes=mm10-filtered-ensembl.gtf
# 得到的结果
ls hg19_and_mm10
fasta/ genes/ pickle/ reference.json star/
如果要增加基因信息
参考链接:https://kb.10xgenomics.com/hc/en-us/articles/115003327112-How-can-we-add-genes-to-a-reference-package-for-Cell-Ranger-
第一步,在fasta/genome.fa的FASTA基础上增加序列信息;
第二步,在genes/genes.gtf的GTF基础上增加注释信息,注意格式
# 每一行有9列tab分隔信息
# 第一列:Chromosome 指定基因组上染色体或contig位置
# 第二列:Source 这个用处不大
# 第三列:Feature CellRanger软件只取exon的部分
# 第四列:Start 起始位点(1-based)
# 第五列:End 终止位点(1-based)
# 第六列:Score 这个用处不大,建议用"."表示
# 第七列:Strand feature信息在基因组的+或-链
# 第八列:Frame 用处不大,建议“.”
# 第九列:分号分隔的键值对,重点是transcript_id 和gene_id。gene_name可选
例如:
mylocus annotation exon 100 200 . + . gene_id "mygene"; transcript_id "mygene";
第三步,使用cellranger mkref运行更新一下
P.S. 最后得到的参考信息(包括参考基因组、注释信息)文件结构如下:
# 这是官网下载的hg38数据
refdata-cellranger-GRCh38-1.2.0
|-- [ 222] README.BEFORE.MODIFYING
|-- [4.0K] fasta
| `-- [2.9G] genome.fa
|-- [4.0K] genes
| `-- [1.3G] genes.gtf
|-- [4.0K] pickle
| `-- [ 58M] genes.pickle
|-- [ 424] reference.json
|-- [4.0K] star
| |-- [3.0G] Genome
| |-- [8.0G] SA
| |-- [1.5G] SAindex
| |-- [1.2K] chrLength.txt
| |-- [1.9K] chrName.txt
| |-- [3.0K] chrNameLength.txt
| |-- [2.1K] chrStart.txt
| |-- [ 37M] exonGeTrInfo.tab
| |-- [ 15M] exonInfo.tab
| |-- [526K] geneInfo.tab
| |-- [ 909] genomeParameters.txt
| |-- [9.1M] sjdbInfo.txt
| |-- [7.1M] sjdbList.fromGTF.out.tab
| |-- [7.1M] sjdbList.out.tab
| `-- [9.4M] transcriptInfo.tab
`-- [ 6] version
4 directories, 21 files
多个文库的整合 aggr
当处理多个生物学样本或者一个样本存在多个重复/文库时,最好的操作就是先分别对每个文库进行单独的count定量,然后将定量结果利用aggr组合起来
第一步 得到count结果
例如现在分别进行3个定量流程
$ cellranger count --id=LV123 ...
... wait for pipeline to finish ...
$ cellranger count --id=LB456 ...
... wait for pipeline to finish ...
$ cellranger count --id=LP789 ...
... wait for pipeline to finish ...
第二步 构建Aggregation CSV
就像这样:
# AGG123_libraries.csv
library_id,molecule_h5
LV123,/opt/runs/LV123/outs/molecule_info.h5
LB456,/opt/runs/LB456/outs/molecule_info.h5
LP789,/opt/runs/LP789/outs/molecule_info.h5
# 其中
# molecule_h5:文件molecule_info.h5 file的路径
第三步 运行aggr
cellranger aggr --id=AGG123 \
--csv=AGG123_libraries.csv \
--normalize=mapped
# 结果输出到AGG123这个目录中
至于最后的 reanalyze ,这个属于定制化分析,这里暂时不做探讨,日后待标准化流程构建起来,再补充这一部分
欢迎关注我们的公众号~_~
我们是两个农转生信的小硕,打造生信星球,想让它成为一个不拽术语、通俗易懂的生信知识平台。需要帮助或提出意见请后台留言或发送邮件到[email protected]
