使用
bowtie/bowtie2进行比对时,一般不建议自己下载基因组构建,建议直接下载索引文件。但是当你需要的参考基因组没有现成的索引文件时,还是需要我们自己构建index,如何建立index呢?
建立index步骤:
- 从UCSC下载参考序列;
- 使用gzip进行解压缩;
- 使用cat将文件进行连接;
- 使用bowtie2-build建立index;
- 使用index
从UCSC下载参考序列
首先进入UCSC Genome browser官网:Downloads ---> Genome Data
点进去后在
Sequence and Annotation Downloads下面有物种的分类,比如,人(human)、哺乳动物(mammals)、昆虫(insects)....等等
假如我们想下载小鼠的参考序列,就从
mammals(哺乳动物)分类里找
mouse后出现如下界面:

我们选择
mm10版本--->
Data set by chromosome 后进入下载界面
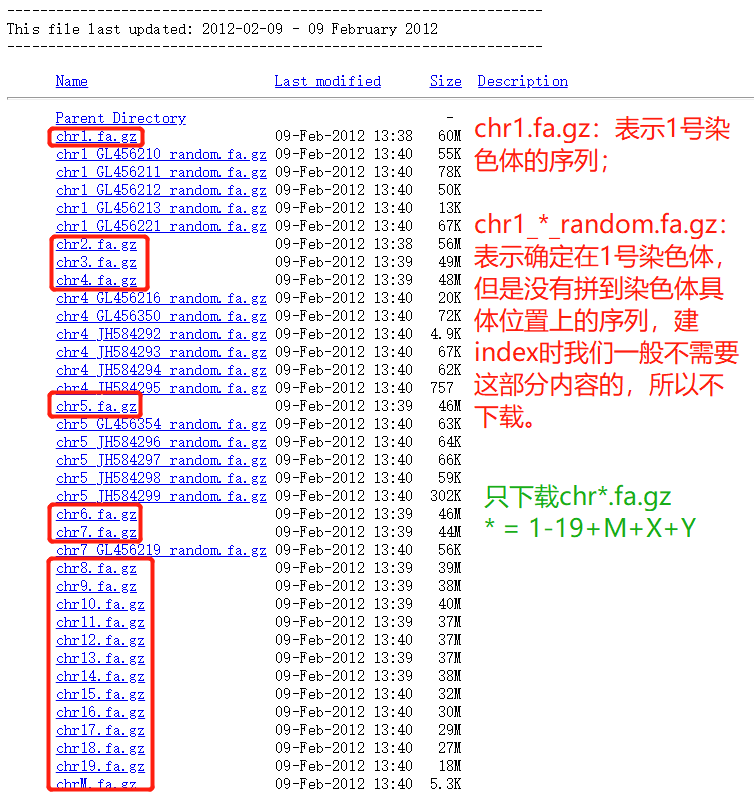
以下载
chr1.fa.gz为例,右击下载界面中
chr1.fa.gz选择
复制链接地址,然后去服务器上运行下载命令
wget:
$ nohup wget -4 -q http://hgdownload.soe.ucsc.edu/goldenPath/mm10/chromosomes/chr1.fa.gz &
下载完成后的格式是:
$ ls -sh
total 61M
61M chr1.fa.gz
检查下载文件大小61M没问题后对chr1.fa.gz解压
使用gzip解压缩
$ gzip -d chr1.fa.gz
解压后chr1.fa就是fasta格式的文件了,参考基因组序列都是fasta格式的
$ ls -sh
total 191M
191M chr1.fa
解压之后我们查看参考基因组1号染色体chr1.fa的前10行:
head -n 10 chr1.fa
$ head -n 10 chr1.fa
>chr1
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
NNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNN
我们可以看到都是N,其实参考基因组中每一条染色体前面有一万个bp都是放N,这是默认的。
使用cat将文件进行连接
按照上述方法,下载并解压好所有需要的参考基因组序列(chr1-19 / chrM / chrX / chrY),我们需要把这22个fa文件进行合并,形成一个大的文件。
正常情况应该这样操作:
$ cat chr1.fa chr2.fa chr3.fa chr4.fa chr5.fa chr6.fa ..... > mm10_genome.fa
#我写省略号是因为我不想写了,实际操作要写完整(chr1-19/chrM/chrX/chrY)
很明显那样写很麻烦,所以我们使用循环来实现:
$ cat merge.sh
list_str=" "
for index in {1..19} X Y M
do
list_str=${list_str}" "chr${index}.fa
done
cat $list_str > mm10_genome.fa
$ nohup bash merge.sh &
有了合并的参考基因组序列,就要用bowtie2-build建立index了。
使用bowtie2-build建立index
bowtie2里的bowtie2-build命令是用来建立index的,我们看下它的核心用法:
Usage: bowtie2-build [options]*
reference_in comma-separated list of files with ref sequences
bt2_index_base write bt2 data to files with this dir/basename
其实就是:bowtie2-build 输入文件(合并好的参考基因组序列) 输出文件名称(自己起)
$ nohup bowtie2-build ./mm10_genome.fa ./mm10 &
超不多1个多小时后就建好index了,建好的index包括6个文件:
$ ll -thr
total 6.7G
-rw-r--r-- 1 jhuang jhuang 633M Mar 21 11:46 mm10.4.bt2
-rw-r--r-- 1 jhuang jhuang 6.0K Mar 21 11:46 mm10.3.bt2
-rw-r--r-- 1 jhuang jhuang 633M Mar 21 12:57 mm10.2.bt2
-rw-r--r-- 1 jhuang jhuang 848M Mar 21 12:57 mm10.1.bt2
-rw-r--r-- 1 jhuang jhuang 633M Mar 21 14:10 mm10.rev.2.bt2
-rw-r--r-- 1 jhuang jhuang 848M Mar 21 14:10 mm10.rev.1.bt2
这时index建立成功。
使用index
在调用index时,我们使用的不是完整的文件的名字,而是使用的这6个文件公共的前缀。
在调用时,我们首先找到index文件所在的路径:
$ pwd
/trainee/huangjing/project/reference/index/bowtie2
在调用index指定文件时,就使用index文件所在路径后面加上前缀mm10:
bowtie2_index = /trainee/huangjing/project/reference/index/bowtie2/mm10
Ref:https://www.bilibili.com/video/av12969326