A survey of best practices for RNA-seq data analysis
RNA-seq数据分析指南
内容
前言
各位同学/老师,大家好,现在由我给大家讲讲我的文献阅读报告!
A survey of best practices for RNA-seq data analysis ,我把它叫做RNA-seq数据分析指南。这篇文章是由佛罗里达大学等单位的研究人员在1月26日发表在Genome Biology上的,该期刊的影响因子有10.8分。这是这篇文章的通讯作者,应该挺靠谱的。
新一代测序技术在爆炸式发展的同时,也衍生出许多其他技术创新。RNA-Seq就是其中之一,这项技术使我们对细胞发育及其调控机制的理解,达到了前所未有的深度和广度。RNA-seq可以获得相当惊人的数据量,而这恰恰是一柄双刃剑。丰富的数据量蕴含着大量的宝贵信息,但这样的数据需要复杂的生物信息学分析,才能从中提取到有意义的结果。
正因如此,数据分析可以说是RNA-seq的重中之重。RNA-seq有非常广泛的应用,但没有哪个分析软件是万能的。科学家们一般会根据自己的研究对象和研究目标,采用不同的数据分析策略。现在人们已经发表了大量的RNA-seq和数据分析方案,对于刚入门的新手来说难免有些无所适从。这篇文章概述了RNA-seq生物信息学分析的现行标准和现有资源,为人们提供了一份RNA-seq数据分析指南,可以作为开展RNA-seq研究的宝贵参考资料。
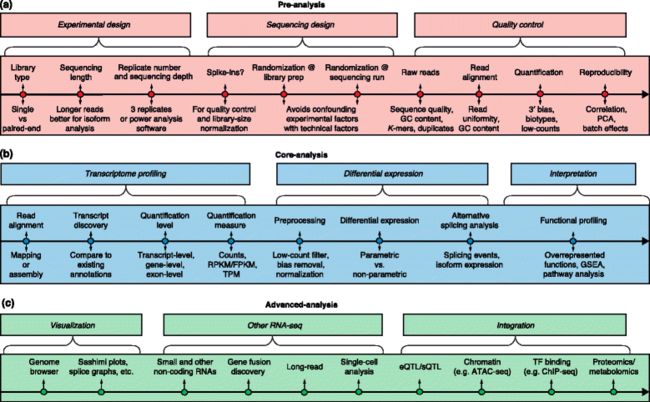
这份指南覆盖了RNA-seq数据分析的所有主要步骤,比如质量控制、读段比对、基因和转录本定量、差异性基因表达、功能分析、基因融合检测、eQTL图谱分析等等。研究人员绘制的RNA-seq分析通用路线图(标准Illumina测序),将主要分析步骤分为前期分析、核心分析和高级分析三类。前期预处理包括实验设计、测序设计和质量控制。核心分析包括转录组图谱分析、差异基因表达和功能分析。高级分析包括可视化、其他RNA-seq技术和数据整合。研究人员在文章中探讨了每个步骤所面临的挑战,也评估了一些数据处理方法的潜力和局限性。此外,他们还介绍了RNA-seq数据与其他数据类型的整合,将基因表达调控与分子生理学和功能基因组学关联起来,这种研究方式如今越来越受到研究者的欢迎。这篇文章在结尾处介绍了一些为转录组领域带来改变的新技术,特别是单细胞RNA-seq和长读段测序技术带来的机遇和挑战。
背景
高通量测序平台
从这张图,横轴是年份,纵轴是测序仪的通量,圈里面的数字代表测序读长。我们可以看到测序仪的通量和读长都在增加了,其中Pacfic Biosciences 的三代测序仪读长最长,可以达到14K,illumina的测序仪通量最大,HiSeq X Ten的通量可以达到1.8T。现在Illumina生产的测序仪占主要的市场份额,我们来单独看看他的测序仪参数。
从左到右,测序仪的通量逐渐增大,它们适合不同的样品和测序目的。Miseq通量比较低,适合宏基因组等微生物测序;Hiseq通量太高了,如果你送去公司测序,他们一般要20~30天才能返回数据,是因为公司要累积很多样品,加标签后一起测序。
高通量测序技术的应用
这幅图的横轴是年份,纵轴是高通量技术应用的代表性文章的引用量。不同的应用技术用颜色进行分类,数据点的大小跟发表率(引用率/月)成正比。可以看出RNA-Seq测序技术的应用最为广泛。
实验设计
RNA-seq到底测的是什么?**
mRNA在生物个体内RNA的组分中只占很小的一部分,rRNA占绝大多数。**一般我们说
RNA-seq指的都是mRNA-seq,后面的流程也都是主要针对mRNA-seq数据分析的。在科学家们的努力下,可以把那些非编码RNA提取出来建库,进行测序。
一个成功的RNA-seq研究,起决定性因素的是一个好的实验设计。还依赖于建库的类型、测序深度和设置适于的生物重复。并且尽量减少测序本身以外带来的数据误差。
文库构建
1.一般生物体中的的RNA中,rRNA占绝大多数,含量超过90%,而mRNA的含量在1-2%左右。对于真核生物,一般使用加poly(A)选择性富集mRNA或者而原核生物则是通过去除rRNA;
2.是否建stand-preserving库;
3.对于Illumina,测序插入片段一般小于500bp。确定合适长度的插入片段是后续测序和分析的关键;
4.单端还是双端测序毫无疑问的是,单端测序更便宜一些,如果你研究的某个物种的基因表达水平,并且它的转录组已经被注释很好了,单端测序产生的数据量一般是足够的了。
双端测序呢,它的读长更长,更适合于那些没有被注释的转录组物种的研究,便于其转录本的从头拼接。
测序深度和重复数
这里的测序深度和重复数的设置呢。
- 测序深度和数据量不能一味加大。测序深度虽然可以提高基因定量和检测的敏感性(低丰度的基因),但是同时也会增加一些噪音和一些无用的转录本。
2.重复数,很重要。(举例子:比较两个班的语文成绩,抽取一个人代表一个班来比较,显然是不科学的。)增加重复数可以减少实验误差,对提高结果的可靠性,是非常有意义的。
误差分为技术误差和生物学差异。
技术误差-可以通过选择最优化的实验测序程序;生物误差-三个生物学重复是最基本的啦。
然后呢,设定生物学重复对差异基因的检出率(真阳性率 TPR)的提高具有明显效果。上面说增加测序深度可以检测到低丰度基因,但是对任何样品来说的当测序深度增加再增加,它就会到达平台期。
由于科研经费有限,无法无限制地增加样本数或数据量。
所以在生物学重复数和单个样本测序量上必须找到平衡点。在总数据量不变的情况下,将总数据量分配到更多的生物学重复样本中,差异分析结果的可靠性在不断提升。
对于RNA-seq,生物学重复数的价值要大于单个样本测序量。但增加生物学重复的样本数,意味着要增加建库费用。因此,即使总数据不变,设置过多的生物学重复也是不合理的。
我们最终确定设置多少生物学重复还是需要看样本个体之间的差异大不大,这点我们一般都很清楚,在测序之前,如果你所研究的现象在两个实验样本之间差异很稳定的话,就可以少设置一些重复,差异不稳定的话有时候设置10个/20个都不够。具体问题具体分析!!
3. 测序深度(Sequencing depth),也叫乘数,指每个碱基被测序的平均次数,是用来衡量测序量的首要参数。研究表明,增加测序深度,测序量从1.6M条reads增加到20M条reads,(75bp)但到10M条reads时就已经达到平衡了,80%的鸡转录本被检测到。在此基础上增加测序量,它们会比对到已经存在的转录本上。
因此即使提高测序深度,低表达水平的基因的检测是比较困难的。并且提高测序深度确实能够增加基因差异表达的敏感度,但是并不能保证检测到的差异具有生物学意义。
这篇2012年BMC Genomics的方法学文章,影响因子3.98至今被引用次数 87次。
直观一些说,如果某个基因在RNA-seq结果显示差异表达,但QPCR结果表明这个基因表达差异不显著,
可以认为这个基因RNA-seq结果为假阳性;反之,这个结果就是真阳性。
生物学重复对差异表达分析的影响
如图所示,在单样本测序量保持不变的情况下,随着生物学重复(n)的提高,差异分析的假阳性率(FPR)基本稳定,但真阳性率(TPR)在不断提高。也就是说提高生物学重复数,实验对差异基因的检测更加敏感,那些差异倍数较小或差异量较低的差异表达基因(此类基因的差异检测难度较大)能够更加容易被检测到。
如表2、3所示,在一定的生物学重复数( n)的情况下,随着单样本测序量(Depth)的提高(25% → 100%),真阳性率(TPR)都只有有限的提高。例如在n=3的情况下,单个样本的测序量从25%提高到100%,TPR仅仅从6.24%提高到8.95%。在表3中,如果Depth等于25%不变,当n从2提高到12,TPR的提高则是非常明显的。因此测序深度对结果改善效果不如增加生物学重复。
总数据量不变,生物学重复数与单样品测序量最佳组合
如果保持总测序量不变(即如果生物量重复数为n,则单个样品的测序量降低为1/n,总数据量为n*1/n=1,保持不变)。如图A,灰色实线代表不同的生物学重复数(n)和单样本数据量(1/n)组合的情况下,真阳性率(TPR)的变化。结果表明,随着n的提高,TPR率不断提高。例如n=2,TPR约为3%,如果n=6,TPR则提高到22%。
同时我们也可以对“单样本测序量对差异表达分析的影响”再进行深入观察。
如果n保持不变,但单个样本的数据量不断降低,TPR的降低十分缓慢。例如,n=3,单个样本的数据量从100%降低到15%,TPR的值一直处于平台期,仅仅从9%降低到5%。 但是不同的生物学重复数和单样本测序量的组合,对假阳性率( FPR)的影响却较小。如图 B,灰色实线代表不同生物学重复数(n)和单样本数据量(1/n)组合的情况下,真阳性率(FPR)的变化。虽然 n 从2 变化到 96,FPR 基本没有太大变化。
从图中我们很容易发现,基于负二项分布的差异分析检验(P value),FPR 对生物学重复数和单个样本数据量均不敏感,始终保持低于 0.1%水平。或者说,这个算法对 FPR 的控制还是非常理想的。
讨论
随着测序单价的下降,目前市场上 RNA-seq 类项目的单样本测序量正在不断提高。以 2G,PE100 测序的表达谱项目为例,其对应的测序量为 20M 条 reads。如果一条长度为 1kbp 的低表达基因的表达量为 RPKM=0.5,其理论上可以检测到的 reads 数为 20×0.5=10。所以低丰度基因的检测,对 RNA-seq 这个技术来说并非最大问题。
第二个问题“转录本表达量的高低变化”比“转录本的有无”更具有普遍的生物学意义。虽然个别基因的表达量变化程度,可以使用 Qpcr 来验证。但我们往往也使用所有差异基因来统计某些规律。例如使用差异基因的 pathway 富集分析来寻找与性状相关的 pathway。如果在全局水平的差异基因集并不可靠,那么 pathway富集分析得出的结论的可靠性自然也受到影响。而全局水平的差异基因数量巨大,是难以使用 Qpcr 验证的。因此,定量以及差异分析的准确性是在 RNA-seq 中更值得关心的问题。
测序设计
RNA-seq文库的制备和测序过程:RNA碎裂,cDNA合成,接头连接,PCR扩增,加标签(多样品混合测序),上泳池测序;
如何减少误差:
1. 使用末端带随机核酸的接头或者使用化学碎裂法代替RNAse III碎裂法;
2. 不同批次实验或者不同runs。
a. 如果样品太多在一个批次或者一个run跑不完,为了避免技术误差造成太大的实验误差,要把样品随机分配到每个批次或runs中;(到底怎么设计,我们要讨论一下!!)
b. 如果你的样品是多样品混合测序,每个样品要单独加上标签,每个lanes要保证足够的测序深度,为了保证所有的样品在每个lane中都有。如果送给公司去做的话,我们要选择建库水平好些的,并且要求他们这么去做,应该会更好。
质量控制
重复数:
技术重复(spearman秩相关系数R²>0.9);生物重复(主成分分析PCA)
原始数据的质量控制:
原始数据回来后,你做完备份以后,做的第一件事情就是看看数据质量如何,一般来自llumina测序平台用软件FastQC看;其他平台的数据用软件NGSQC。一般会有原始数据的序列质量,GC含量,存在的接头以及K-mers子串图并且重复序列太多的reads。
并且reads 3‘末端的质量低于前段,原因是随着测序读长的增加,酶活性下降,荧光强度也在下降,因此测序数据质量逐渐降低乃是自然趋势。常用的数据过滤的软件有FASTX-Toolkit and Trimmomatic,其他还有许多,你也可以自己写代码处理数据。
Reads比对后的质量控制(评估比对质量的指标):**比对上的reads占总reads的百分比; Reads比对到外显子和参考链上的覆盖度是否一致;比对到基因组序列:多重比对reads?比对到转录组序列:来自未被注释的转录本的reads会丢失; 产生更多的多重比对reads; 转录本被定量以后,应该看一下GC含量和基因长度偏差,确定定量的方法是否适用。
转录本分析
把所有样本的reads混合用于转录本的拼接。二代测序的转录组reads用于拼接还是存在一些问题的,最终拼接结果不太理想。一个转录本的拼接结果会是10~100contigs。三代测序的读长直接可以把一个转录本读完了,完全不需要拼接。
RPKM/FPKM/TPM用来表示RNA-seq基因表达水平的值;对于单端测序RPKM和FPKM值是一样的,FPKM可以转换成TPM。Cufflinks(支持双端测序数据,并且需要GTF格式的注释文件)定量算法有……
提一个问题,有那么多软件到底怎样才是好的,选哪个软件好呢?
功能分析
功能分析是标准转录组分析流程的最后一步,分析差异表达基因的分子功能和代谢通路。
其他RNA-seq应用
小RNA:
1.小RNA的长度通常在18~34个碱基,包含了miRNAs, short-interfering RNAs (siRNAs),PIWI-interactingRNAs (piRNAs)以及其他种类的**。
- sRNA-seq libraries are rarely sequenced as deeply as regular RNA-seq libraries because of a lack of complexity, with a typical range of 2–10 million reads.
3. 小RNA的数据分析流程跟常规RNA的分析流程不同。
- miRTools 2.0 ,
a tool for prediction and profiling of sRNA species, uses by default reads that are 18–30 bases long
5. 比对到参考基因组上,比对软件有:
Bowtie2 ,STAR , or Burrows-Wheeler Aligner (BWA) PatMaN and MicroRazerS map short sequences
多种数据整合分析
1. Moreover, the combination of RNA-seq and re-sequencing can be used both to remove false positives when inferring fusion genes and to analyze copy number alterations.
2. The statistically significant correlations that were observed, however, accounted for relatively small effects. (DNA methylation)
3. ….
4. 一些分析软件:CORNA, MMIA,, MAGIA, and SePIA;
5.代谢组和转录组数据结合进行通路分析,有一些软件:MassTRIX, Paintomics, VANTED v2, and SteinerNet
整合多种组学数据分析还不是很成熟,但是仍有一些软件可以用。
展望
RNA-seq技术已经成为转录组分析的标准方法。其相对应的技术和数据分析工具还在不断地发展。
对低表达的基因的定量仍是一个等待解决的问题; 三代测序技术,Smart-seq和Smart-seq2应用于转录组测序,所需要的样品量少,并且可以测定单细胞内的RNA表达水平; Pacbio 技术可以直接测得接近全长的转录本,可以有效解决二代测序技术拼接较为零碎以及潜在嵌合拼接的问题;
目前的瓶颈:价格高(建库价格和测序价格);
(1)需要多种长度的文库;
(2)测序通量有限;Pacbio新推出的sequel测序仪,比旧版本测序仪,通量提高了7倍(测序芯片的波导空数量从15万,提升到100万。所以,有望进一步提高Pacbio在转录组De Novo中的应用面。
PS: 综述类的文章主要是讲解原理和优缺点,以及当前趋势和建议,不可能说看个综述就学会了RNA-seq的全套分析,但是不看综述,不了解原理,所有的分析却都是无源之水无本之木。
还有,综述给的建议,包括样本数,数据量这些东西都是实验设计之初就得考虑的,但是大多数生信工程师拿到数据很多东西就没得改变了,所以总是有人问如果没有重复的转录组数据用什么R包来找差异这样的问题。请大家分清楚理想和现实的差别。
摘要:
佛罗里达大学、加州大学Irvine分校等单位的研究人员在一月二十六日的Genome Biology杂志上发表文章,概述了RNA-seq生物信息学分析的现行标准和现有资源,为人们提供了一份带有注释的RNA-seq数据分析指南。这将成为开展RNA-seq研究的宝贵参考资料。
生物通报道:新一代测序技术在爆炸式发展的同时,也衍生出许多其他技术创新。RNA深度测序(RNA-Seq)就是其中之一,这项技术使我们对细胞发育及其调控机制的理解,达到了前所未有的深度和广度。尽管研究细胞RNA并不是什么新鲜事,但RNA-Seq的出现大大拓展了转录组研究的规模,取得了累累硕果,这些是传统技术难以企及的。
RNA-seq可以获得相当惊人的数据量,而这恰恰是一柄双刃剑。丰富的数据量蕴含着大量的宝贵信息,但这样的数据需要复杂的生物信息学分析,才能从中提取到有意义的结果。正因如此,数据分析可以说是RNA-seq的重中之重。
RNA-seq有非常广泛的应用,但没有哪个分析软件是万能的。科学家们一般会根据自己的研究对象和研究目标,采用不同的数据分析策略。现在人们已经发表了大量的RNA-seq和数据分析方案,对于刚入门的新手来说难免有些无所适从。
佛罗里达大学、加州大学Irvine(****尔湾)分校等单位的研究人员在一月二十六日的Genome Biology杂志上发表文章,概述了RNA-seq生物信息学分析的现行标准和现有资源,为人们提供了一份带有注释的RNA-seq数据分析指南。这将成为开展RNA-seq研究的宝贵参考资料。
这份指南覆盖了RNA-seq数据分析的所有主要步骤,比如质量控制、读段比对、基因和转录本定量、差异性基因表达、功能分析、基因融合检测、eQTL图谱分析等等。研究人员绘制的RNA-seq分析通用路线图(标准Illumina测序),将主要分析步骤分为前期分析、核心分析和高级分析三类。前期预处理包括实验设计、测序设计和质量控制。核心分析包括转录组图谱分析、差异基因表达和功能分析。高级分析包括可视化、其他RNA-seq技术和数据整合。
研究人员在文章中探讨了每个步骤所面临的挑战,也评估了一些数据处理方法的潜力和局限。此外,他们还介绍了RNA-seq数据与其他数据类型的整合。这种数据整合可以将基因表达调控与分子生理学和功能基因组学关联起来,如今越来越受到研究者的欢迎。
这篇文章在结尾处介绍了一些为转录组领域带来改变的新技术,特别是单细胞RNA-seq和长读取测序技术带来的机遇和挑战。
2015年年初,RNA-Seq的数据分析方法如雨后春笋般涌现。三月份,Nature集团旗下刊物发表了三篇介绍RNA-Seq数据分析新方法的文章,一篇发表在《Nature Methods》上,另外两篇发表在《Nature Biotechnology》上。这三篇文章有一位共同的作者,那就是约翰霍普金斯大学计算生物学中心的Steven Salzberg,生物信息学和计算生物学领域的杰出科学家。Salzberg通过这些文章中分别介绍了三种新工具:HISAT、StringTie和Ballgown。这些工具可以取代之前开发的早期工具,为RNA-Seq提供了全新的数据分析方法,从原始数据读取到差异表达分析。(更多详细信息参见:三篇文章介绍RNA-Seq数据分析的新工具)
RNA测序究竟有多可靠呢?由美国FDA牵头的测序质量控制(SEQC)项目对RNA测序的准确性、可重现性和信息含量进行了综合性评估。其初步调查结果发表在2014年09月的Nature Biotechnology杂志上,石乐明教授是这篇文章的通讯作者之一。研究人员用RNA参照样本在全球多个实验室的Illumina HiSeq、Life Technologies SOLiD、Roche 454平台上进行检测,主要评估RNA测序在接头区域和差异性表达谱中的表现,并将其与芯片和定量PCR(qPCR)进行比较。研究表明,数据分析的算法会对RNA测序产生很大影响,不同算法生成的转录本数据存在很大差异。(更多详细信息参见:石乐明教授Nature子刊:RNA测序到底可不可靠)
前几天,浙江大学和哈佛大学的研究人员在Cell Reports杂志上发表了一项单细胞mRNA-seq研究。基因表达变异是小鼠胚胎干细胞(ESC)的一个重要特征,但人们一直不清楚这背后的具体原因。研究人员通过分析小鼠胚胎干细胞发现,这些细胞表现出的异质性是血清培养造成的。他们在其中鉴定了高度变异的基因簇,以及独特的染色质状态。研究显示,双价基因(bivalent gene)更容易出现表达变异。进一步研究表明,无血清培养可以减少小鼠ESC的异质性和转录组变异。这意味着,细胞内的网络变异大多是细胞外的培养环境造成的。(更多详细信息参见:浙大80后教授发表单细胞RNA测序研究)