linux基本命令
第六章 Linux文件与目录管理
极客学院服务器
高通量相关基础知识
生信一百讲
1、 Anaconda
Anaconda是一个Python下和Canopy类似的的科学计算环境,但用起来更加方便。自带的包管理器conda也很强大,可以解决大多数软件的安装和配置
-
1-1、conda的安装
Conda 官网 https://conda.io/docs/index.html
mkdir bio_soft ## 创建一个文件夹,主演们用来放置软件
cd bio_soft/ ## 进入到该文件夹目录下面
wget https://repo.continuum.io/archive/Anaconda2-5.1.0-Linux-x86_64.sh ## 下载python2.7的Anaconda
mkdir conda
mv Anaconda2-5.1.0-Linux-x86_64.sh conda ## 将所下载的文件移动到conda目录下
cd conda
bash Anaconda2-5.1.0-Linux-x86_64.sh ## 运行bash命令,一路回车或者yes,会自动在用户所属环境创建一个anaconda2的文件夹,并且自动添加环境变量
export PATH=/home/u883604/anaconda2/bin:$PATH ## 添加路径到环境变量(已添加,可省略)
source ~/.bashrc
conda ##检测是否安装成功
添加几个通道
conda config --add channels r
conda config --add channels defaults
conda config --add channels conda-forge
conda config --add channels bioconda
无论是conda默认的软件源还是bioconda软件源都是国外的,速度非常慢,所以需要增加国内软件源,同时bioconda已经有清华,中科大两个国内镜像,也添加进去
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --set show_channel_urls yes ## 设置搜索时显示通道地址
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/msys2/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/
查看目前conda软件源情况
conda info
-
1-2、 Conda的几个常用命令
conda search PACKAGENAME ## 查找是否有所需的安装文件
## 也可以在线查询 https://anaconda.org/
conda install PACKAGENAME ## 安装所需软件
## conda默认安装最新版本的软件,若想安装非最新版的软件输入:conda search bwa查看可选版本
conda install bwa=版本号
conda list ## 查看所有安装的软件
conda update 软件名 可以对软件进行升级:eg. conda update bwa
conda remove 卸载已经安装的软件
更多详细命令
conda commands
-
1-3 、 Conda环境管理
## 由于我们下载的是python2.7版本的,但是如果我们需要运行python3.0以上的呢?
# 创建一个名为python3的环境,指定Python版本是3.6
conda create --name python3 python=3.6
source activate python3 ## 加载python3的环境
source deactivate ## 使用结束,可以退出环境
conda remove --name python34 --all ## 删除一个已有的环境
参考链接
生信札记
PeterYuan
2、 RNA-seq软件安装
- Tophat2
conda 直接安装
conda install -c bioconda sra-tools
conda install fastqc ## 不知道是网速还是怎么下载中断好几次,所以改为手动安装了
conda install trimmomatic
conda install tophat2
conda install bowtie2
conda install samtools
conda install cufflinks
有些软件用conda 无法安装,可以尝试手动安装

预编译版本安装
cd bio_soft
[tophat2官网](http://ccb.jhu.edu/software/tophat/index.shtml)
wget http://ccb.jhu.edu/software/tophat/downloads/tophat-2.1.1.Linux_x86_64.tar.gz ## 下载已经编译好了的
mv tophat-2.1.1.Linux_x86_64/ tophat2
cd tophat2/
## 添加环境变量
## 临时添加环境变量
export PATH=/home/u883604/bio_soft/tophat2/:$PATH
## 长久使用需要修改 ~/.bashrc 文件
vim ~/.bashrc
PATH=/home/u883604/bio_soft/tophat2/:$PATH ## tophat2所在路径
source ~/.bashrc
tophat2 -h
源代码安装
如果以上方法安装不成功,可以使用源文件(文件名通常含src)的版本安装
wget 下载链接
tar -zxvf 文件名
cd 文件
#看readme文件里的安装提示,没有该文件的话,自己网上查找
解压命令:
tar -zxvf xxx.tar.gz
tar -jxvf xxx.tar.bz2
安装分三步:
1. 配置: ./configure --prefix=想要指定的路径
2. 编译: make
3. 安装: make install
若安装出问题(没有权限在系统目录下安装),需要清空上次编译的文档
make clean
./configure --prefix=安装路径
make
make insatll #可执行文件通常在bin目录下,环境变量设置和源码安装一样
FastQC安装
wget http://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.7.zip
unzip fastqc_v0.11.7.zip
cd FastQC/
chmod 777 fastqc
./fastqc -h
vim ~/.bashrc
export PATH=/home/u883604/bio_soft/FastQC/:$PATH
source ~/.bashrc
cufflinks安装
wget http://cole-trapnell-lab.github.io/cufflinks/assets/downloads/cufflinks-2.2.1.Linux_x86_64.tar.gz
tar -xzvf cufflinks-2.2.1.Linux_x86_64.tar.gz
cd cufflinks-2.2.1.Linux_x86_64/
vim ~/.bashrc
export PATH=/home/u883604/bio_soft/cufflinks-2.2.1.Linux_x86_64/:$PATH
source ~/.bashrc
3、 RNA-seq分析流程
3.1下载水稻的参考基因组文件和注释文件
水稻数据库
mkdir -p rna_practice/ref 创建
nohup wget http://rice.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/all.con & ## 参考基因组
nohup wget http://rice.plantbiology.msu.edu/pub/data/Eukaryotic_Projects/o_sativa/annotation_dbs/pseudomolecules/version_7.0/all.dir/all.gff3 & ## 注释文件
# 使用nohup &可以将任务放置后台运行
下载拟南芥的参考基因组文件和注释文件
## 创建文件夹用来放置参考基因组或注释文件
mkdir -p rna_practice/data
cd rna_practice/ref
nohup wget ftp://ftp.ensemblgenomes.org/pub/plants/release-28/fasta/arabidopsis_thaliana/cdna/Arabidopsis_thaliana.TAIR10.28.cdna.all.fa.gz &
nohup wget ftp://ftp.ensemblgenomes.org/pub/plants/release-28/fasta/arabidopsis_thaliana/dna/Arabidopsis_thaliana.TAIR10.28.dna.genome.fa.gz &
nohup wget ftp://ftp.ensemblgenomes.org/pub/plants/release-28/gff3/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.28.gff3.gz &
nohup wget ftp://ftp.ensemblgenomes.org/pub/plants/release-28/gtf/arabidopsis_thaliana/Arabidopsis_thaliana.TAIR10.28.gtf.gz &
# 解压缩
gzip -d *.gz
下载测序数据
# 创建一个文件夹,用来存放FASTQ文件(或者公司返回的数据)
mkdir -p rna_practice/data/
cd rna_practice/data/
# 获取样本信息
wget http://www.ebi.ac.uk/arrayexpress/files/E-MTAB-5130/E-MTAB-5130.sdrf.txt
tail -n +2 E-MTAB-5130.sdrf.txt | cut -f 32,36 |sort -u
# 下载数据
# fastq文件下载链接在第几列
# head -n1 E-MTAB-5130.sdrf.txt | tr '\t' '\n' | nl | grep URI
# 根据上述返回数字,获取文件第33列,然后下载fastq文件
# tail -n +2 E-MTAB-5130.sdrf.txt | cut -f 33 | xargs -i wget {}
# 也可以编写批量下载数据的shell脚本,如下:
perl -alne 'if(/.*(ftp:.*gz).*/){print "nohup wget $1 &"}' E-MTAB-5130.sdrf.txt >fq_data_download.sh
bash fq_data_download.sh

下载测序数据
# 创建一个文件夹,用来存放FASTQ文件(或者公司返回的数据)
mkdir -p rna_practice/data/
cd rna_practice/data/
#第一种方法
nohup prefetch -v SRR1999345 &
nohup prefetch -v SRR1999346 &
nohup prefetch -v SRR1999347 &
nohup prefetch -v SRR1999348 &
#第二种方法
cat SRR_Acc_List.txt | xargs prefetch -v
#第三种方法
### 创建一个down.sh文件,里面内容如下:
#!/bin/bash
for i in `seq 5 8`
do
prefetch -v SRR199934${i} &
bash down.sh
本次数据
## ftp格式为SRR/SRR+SRR数字的前三位/SRR号/SRR号.sra
nohup wget ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR350/SRR3503149/SRR3503149.sra &
nohup wget ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR350/SRR3503154/SRR3503154.sra &
nohup wget ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR350/SRR3503151/SRR3503151.sra &
nohup wget ftp://ftp.ncbi.nlm.nih.gov/sra/sra-instant/reads/ByRun/sra/SRR/SRR350/SRR3503152/SRR3503152.sra &
将sra文件转换为fq文件
第一种:创建一个fastq-dump.sh文件,内容如下
#!/bin/bash
for i in `seq 5 9`
do
nohup fastq-dump --split-3 --gzip SRR199934${i}.sra
done&
nohup bash fastq-dump.sh &
第二种:创建一个fastq-dump.sh文件,内容如下
#PBS -N fastq-dump ## 指定任务名 (自定义)
#PBS -l nodes=1:ppn=4 ## 1表示一个线程 4表示4个核 同时也可以指定某个节点如 nodes=node32:ppn=4 (一般不需要改,4个基本够)
#PBS Cl walltime=1:00:00 //请求任务执行时间
#PBS -q batch
#PBS -V
cd $PBS_O_WORKDIR
ls *.sra | xargs fastq-dump --split-3 --gzip ## 所执行的命令(自定义)
qsub fastq-dump.sh ##提交任务到服务器
集群使用命令参考:
http://hpc.ncpgr.cn:8093/mediawiki/index.php/PBS%E4%BD%BF%E7%94%A8


本流程将sra文件转换为fastq文件使用命令
fastq-dump 一般使用参数
## 单端数据使用命令
fastq-dump SRR***.sra -O out_path
fastq-dump --fasta SRR306998.sra -O out_path
## 双端数据使用命令
fastq-dump --split-3 SRR***.sra -O out_path
## 在这里我们是采用的是双端测序的数据,所以使用双端的命令
fastq-dump --split-3 --gzip Cr-DJ-2-1.sra # --gzip 是指生成的文件为压缩的文件,可以节省占用内存
fastq-dump --split-3 --gzip Cr-DJ-2-2.sra
fastq-dump --split-3 --gzip osdrm2-1.sra
fastq-dump --split-3 --gzip osdrm2-2.sra
## 会得到8个fastq文件
得到了fastq文件我们就可以采用不同的RNA-seq protocol来进行分析了
- hppRNA—a Snakemake-based handy parameter-free pipeline for RNA-Seq analysis of numerous samples

在进行数据分析前必须进行数据质量的检测,今天略过。。。。
Tophat –> Cufflink –> Cuffdiff
1、 对基因组序列建立索引
## 特别注意这里使用的index的gtf或者gff文件一定要与基因组序列文件要对应
mkdir rice_bowtie2_index ## 存放水稻参考基因组文件
cd rice_bowtie2_index
bowtie2-build all.fa rice ## rice为输出的索引的前缀
mkdir rice_gtf ## 存放水稻的注释文件即gff3文件或者gtf文件
mkdir data
cd data
ln -s /public/home/qliu/data/B512_data/RNA_data/osdrm2/*.fastq ./ ##建立软链接(类似windows里面的快捷键)
2、将测序数据的reads比对到参考基因组上
mkdir align
cd align
## 这里使用的命令皆为首先创建sh文件,然后提交到集群运行,具体格式参考之前
tophat -p 8 -o CrDJ-1 -G /public/home/qliu/RNA_practice/rice_gtf/all.gff3 /public/home/qliu/RNA_practice/rice_bowtie2_index/rice /public/home/qliu/RNA_practice/data/Cr-DJ-2-1_1.fastq /public/home/qliu/RNA_practice/data/Cr-DJ-2-1_2.fastq
tophat -p 8 -o CrDJ-2 -G /public/home/qliu/RNA_practice/rice_gtf/all.gff3 /public/home/qliu/RNA_practice/rice_bowtie2_index/rice /public/home/qliu/RNA_practice/data/Cr-DJ-2-2_1.fastq /public/home/qliu/RNA_practice/data/Cr-DJ-2-2_2.fastq
tophat -p 8 -o osdrm-1 -G /public/home/qliu/RNA_practice/rice_gtf/all.gff3 /public/home/qliu/RNA_practice/rice_bowtie2_index/rice /public/home/qliu/RNA_practice/data/osdrm2-1_1.fastq /public/home/qliu/RNA_practice/data/osdrm2-1_2.fastq
tophat -p 8 -o osdrm2-2 -G /public/home/qliu/RNA_practice/rice_gtf/all.gff3 /public/home/qliu/RNA_practice/rice_bowtie2_index/rice /public/home/qliu/RNA_practice/data/osdrm2-2_1.fastq /public/home/qliu/RNA_practice/data/osdrm2-2_2.fastq
## 参数说明
-p 8 表示使用8个线程
-o CrDJ-1 表示结果文件输出到CrDJ-1文件夹中(不需要自己提前创建)
-G 后面跟基因组注释文件
/public/home/qliu/RNA_practice/rice_bowtie2_index/rice 表示之前所建立的基因组index所指定的前缀
所以参考的模板为
tophat -p 8 -G genes.gtf -o C1_R1_thout index C1_R1_1.fq C1_R1_2.fq
## 注意如果是链特异性测序比对的时候需要加参数 --library-type (后跟你的测序类型)
fr-firststrand,fr-secondstrand
(详情见 https://www.jianshu.com/p/a63595a41bed )
3、对每个样本进行转录本组装
mkdir cufflinks
cufflinks -p 8 -o CrDJ-1 /public/home/qliu/RNA_practice/align/CrDJ-1/accepted_hits.bam
cufflinks -p 8 -o CrDJ-2 /public/home/qliu/RNA_practice/align/CrDJ-2/accepted_hits.bam
cufflinks -p 8 -o osdrm2-1 /public/home/qliu/RNA_practice/align/osdrm-1/accepted_hits.bam
cufflinks -p 8 -o osdrm2-2 /public/home/qliu/RNA_practice/align/osdrm2-2/accepted_hits.bam
4、将所有的转录本进行组装
- 首先创建一个assemblies.txt(里面应包含上一部得到的gtf文件的路径)
## 创建文件可以使用vim assemblies.txt (将gft文件所在路径粘贴进去即可)
/public/home/qliu/RNA_practice/cufflinks/CrDJ-1/transcripts.gtf
/public/home/qliu/RNA_practice/cufflinks/CrDJ-2/transcripts.gtf
/public/home/qliu/RNA_practice/cufflinks/osdrm2-1/transcripts.gtf
/public/home/qliu/RNA_practice/cufflinks/osdrm2-1/transcripts.gtf
- 将所有的转录本合并到一个转录本中,形成一个新的转录本注释文件
## 不需要提交服务器 直接运行即可
mkdir cuffmerge
cd cuffmerge
cuffmerge -g /public/home/qliu/RNA_practice/rice_gtf/all.gff3 -s /public/home/qliu/RNA_practice/rice_bowtie2_index/all.fa -p 8 assemblies.txt
参数说明
-g 后面跟参考基因组的注释文件
-s 后面跟参考基因组序列文件
-p 线程数
5、获得基因表达文件
mkdir cuffdiff
cuffdiff -o ./diff_out/ -b /public/home/qliu/RNA_practice/rice_bowtie2_index/all.fa -p 8 -L CrDJ,osdrm2 -u /public/home/qliu/RNA_practice/cuffmerge/merged_asm/merged.gtf /public/home/qliu/RNA_practice/align/CrDJ-1/accepted_hits.bam,/public/home/qliu/RNA_practice/align/CrDJ-2/accepted_hits.bam /public/home/qliu/RNA_practice/align/osdrm-1/accepted_hits.bam,/public/home/qliu/RNA_practice/align/osdrm2-2/accepted_hits.bam
6、根据文章设立的阈值,挑选差异基因
awk '{if(($10<-2)&&($11<0.001))print $3"\t"$8"\t"$9"\t"$10}' gene_exp.diff | grep -v 'inf' > down.txt ## 筛选出下调的基因(log2_fold_change < -2 & pvalue < 0.001)
awk '{if(($10>2)&&($11<0.001))print $3"\t"$8"\t"$9"\t"$10}' gene_exp.diff | grep -v 'inf' > up.txt ## 筛选出上调的基因(log2_fold_change > 2 & pvalue < 0.001
## 参数说明
grep -v 输出不含inf的所有信息
Subread -> featureCounts -> DESeq2
1、 subread-buildindex 建立索引 (速度贼快 ~2.4min,可在计算节点直接运行,不需提交)
subread-buildindex -o rice rice.fa
# rice.fa 为之前一样的水稻参考基因组序列
# -o 输出索引的前缀
2、 subjunc 进行比对
mkdkir align
cd align
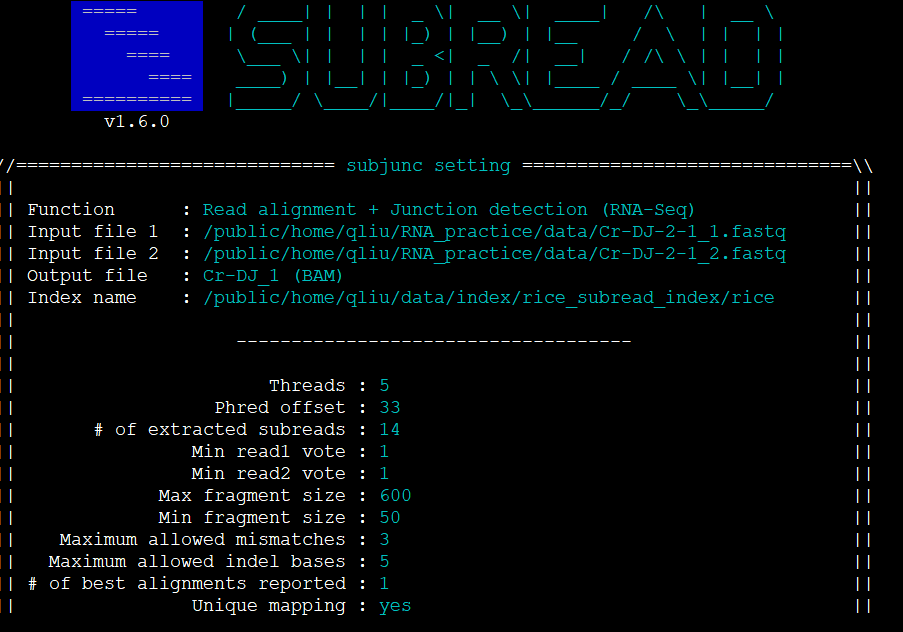
subjunc -T 5 -i /public/home/qliu/data/index/rice_subread_index/rice -r /public/home/qliu/RNA_practice/data/Cr-DJ-2-1_1.fastq -R /public/home/qliu/RNA_practice/data/Cr-DJ-2-1_2.fastq -o Cr-DJ_1
subjunc -T 5 -i /public/home/qliu/data/index/rice_subread_index/rice -r /public/home/qliu/RNA_practice/data/Cr-DJ-2-2_1.fastq -R /public/home/qliu/RNA_practice/data/Cr-DJ-2-2_2.fastq -o Cr-DJ_2
subjunc -T 5 -i /public/home/qliu/data/index/rice_subread_index/rice -r /public/home/qliu/RNA_practice/data/osdrm2-1_1.fastq -R /public/home/qliu/RNA_practice/data/osdrm2-1_2.fastq -o osdrm2_1
subjunc -T 5 -i /public/home/qliu/data/index/rice_subread_index/rice -r /public/home/qliu/RNA_practice/data/osdrm2-2_1.fastq -R /public/home/qliu/RNA_practice/data/osdrm2-2_2.fastq -o osdrm2_2
## 查看bam文件命令,比如查看前几行
samtools view file.bam |head
## 参数说明 (具体说明直接运行 subjunc )
-T 线程数
-i 之前建立的基因组索引的前缀
-r 接fastq文件
-R 双端测序的fastq文件
-o 输出名

3、 featureCounts 进行定量
mkdir featureCounts
cd featureCounts
featureCounts -p -T 6 -a /public/home/qliu/RNA_practice/rice_gtf/rice.gtf -o Cr_DJ-osdrm2_fCount.out /public/home/qliu/RNA_practice/subread/align/Cr-DJ_1 /public/home/qliu/RNA_practice/subread/align/Cr-DJ_2 /public/home/qliu/RNA_practice/subread/align/osdrm2_1 /public/home/qliu/RNA_practice/subread/align/osdrm2_2
## 注意参数
-t 默认exon
-g 默认gene_id
用之前请查看自己的gff文件或者gtf文件
4、 提取定量的信息
awk -F '\t' '{print $1,$7,$8,$9,$10}' OFS='\t' Cr_DJ-osdrm2_fCount.out > WT_osdrm2_matrix.out
## Cr_DJ-osdrm2_fCount.out数据中只有第一列和从第七列(即第一个bam文件)到最后一列信息才是我们所需要的,即我们这里有四个bam文件所以提取第7到10列
# 参数说明
\t 表示以制表符分割开来
5、 将矩阵导入R中,采用DESeq2进行差异分析
> countdata <- read.table("WT_osdrm2_matrix.out", header=TRUE, row.names=1) #导入数据
>
> head(countdata) # 查看数据前几行(列名 太长自己展示看)
>
> colnames(countdata) <- c("WT_1","WT_2","DRM2_1","DRN2_2") # 修改列名
> head(countdata)
WT_1 WT_2 DRM2_1 DRN2_2
LOC_Os01g01010 1250 1250 948 574
LOC_Os01g01019 66 66 136 114
LOC_Os01g01030 535 535 665 464
LOC_Os01g01040 1531 1531 1225 847
LOC_Os01g01050 556 556 742 524
LOC_Os01g01060 2925 2925 3312 2374
> dim(countdata) # 查看数据维度,即几行几列
[1] 55986 4
> condition <- factor(c("WT","WT","DRM2","DRM2")) # 赋值因子,即变量
> library(DESeq2) # 这一步以后基本可以复制粘贴
> coldata <- data.frame(row.names=colnames(countdata), condition) # 创建一个condition数据框
> dds <- DESeqDataSetFromMatrix(countData=countdata, colData=coldata, design=~condition) ##构建dds矩阵(后面很多分析都是基于这个dds矩阵)
> dds <- DESeq(dds)
> resdata <- results(dds)
> table(resdata$padj<0.05) # p<0.05的基因数
> res_padj <- resdata[order(resdata$padj), ] ##按照padj列的值排序
> names(resdata)[1] <- "Gene" # 将第一列的列名改为Gene
> write.table(resdata, file="diffexpr_padj_results.csv",sep = "\t",row.names = F) ## 将结果文件保存到本地
## 筛选差异基因
> subset(resdata,pvalue < 0.001) -> diff ## 先筛选pvalue < 0.01的行
> subset(diff,log2FoldChange < -2) -> down ## 筛选出下调的
> subset(diff,log2FoldChange > 2) -> up ## 筛选出上调的
> dim(down)
[1] 1124 11
> dim(up)
[1] 3023 11
6、 DESeq2分析中得到的resdata进行绘制火山图
rm(list=ls())
## 加载DESeq2中生成的resdata文件
resdata <- read.csv(file.choose(),header = T , sep = "\t")
threshold <- as.factor(ifelse(resdata$padj < 0.001 &
abs(resdata$log2FoldChange) >= 2 ,
ifelse(resdata$log2FoldChange >= 2 ,'Up','Down'),'Not'))
ggplot(resdata,aes(x=log2FoldChange,y=-log10(padj),colour=threshold)) +
xlab("log2(Fold Change)")+ylab("-log10(qvalue)") +
geom_point(size = 0.5,alpha=1) +
ylim(0,200) + xlim(-12,12) +
scale_color_manual(values=c("green","grey", "red"))

原文献图

7、 GO富集分析
- 打开网页PCSD输入我们得到的差异基因,这里拿上调的差异基因为例,看到有几个选项,因为我这里是没有区分TE和Non TE的所以我直接选了第一个

- 点击submit
## 由于下载分GOSlim文件的ID为转录本ID,所以要先进行处理
sed -e 's/\.[0-9]\{1\}//g' all.GOSlim_assignment > new_GOSlim.txt
## 将gene ID转换为GO ID
all.GOSlim <- read.table("/public/home/qliu/data/rice_data/rice_all.dir/new_GOSlim.txt",sep = "\t",header =T)
colnames(all.GOSlim) <- c("ID","GO_number","Background","Pathyway","IEA","TAIR")
up_gene <- read.table("up.gene",header=FALSE)
colnames(up_gene) <- "ID"
library(dplyr)
## 筛选出up gene 所在的GO_ID
left_join(up_gene,all.GOSlim,by = "ID") -> temp.data
GO_ID <- as.matrix(temp.data[,2])
GO_ID <- as.character(GO_ID[,1])
require(AnnotationHub)
ah <- AnnotationHub()
ah$species[which(ah$species == "Oryza sativa")] ## 水稻有几个数据库
query(ah, "Oryza sativa") ## 查找编号
> query(ah, "Oryza sativa")
AnnotationHub with 2 records
# snapshotDate(): 2017-10-27
# $dataprovider: Inparanoid8, ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/
# $species: Oryza sativa, Oryza sativa_Japonica_Group
# $rdataclass: Inparanoid8Db, OrgDb
# additional mcols(): taxonomyid, genome, description, coordinate_1_based, maintainer,
# rdatadateadded, preparerclass, tags, rdatapath, sourceurl, sourcetype
# retrieve records with, e.g., 'object[["AH10561"]]'
title
AH10561 | hom.Oryza_sativa.inp8.sqlite
AH59059 | org.Oryza_sativa_Japonica_Group.eg.sqlite
## subset(ah, species == 'Oryza sativa' & rdataclass == 'OrgDb')
rice_ref <- ah[['AH59059']]
## 查看rice_ref信息
str(rice_ref)
mode(rice_ref)
class(rice_ref)
columns(rice_ref) ## 这个可看
keytypes(rice_ref) ## 这个后面需要用到,这里我导入的是的GO号
head(keys(rice_ref,keytype = "GO"))
library("clusterProfiler")
PP_GO <- read.table(file.choose(),header=FALSE) ## 只有GO号的文件
PP_GO <- as.character(PP_GO[,1])
PP_test <- enrichGO(gene = PP_GO,
OrgDb = rice_ref,
keyType = "GO",
pAdjustMethod = "BH", ## “holm”, “hochberg”, “hommel”, “bonferroni”, “BH”, “BY”, “fdr”, “none”
ont = "CC" , ## 可选"BP" "CC" "MF"
pvalueCutoff = 0.01,
qvalueCutoff = 0.05)
dotplot(PP_test, showCategory=30)
enrichMap(PP_test, vertex.label.cex=1.2,layout=igraph::layout.kamada.kawai)
barplot(PP_test,showCategory=12,font.size=7,title="groupGO Cellular Component")
library(topGO)
plotGOgraph(PP_test)

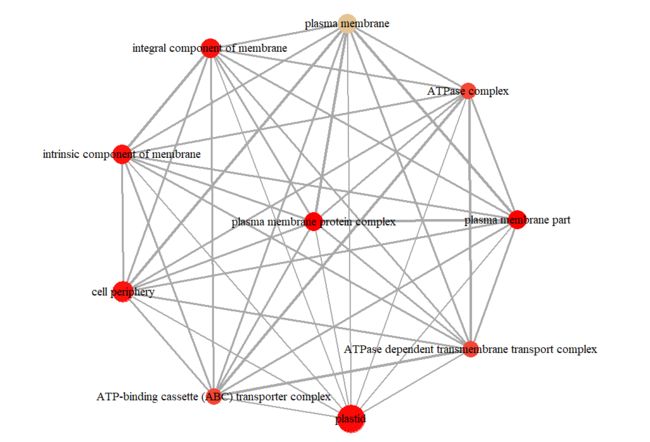
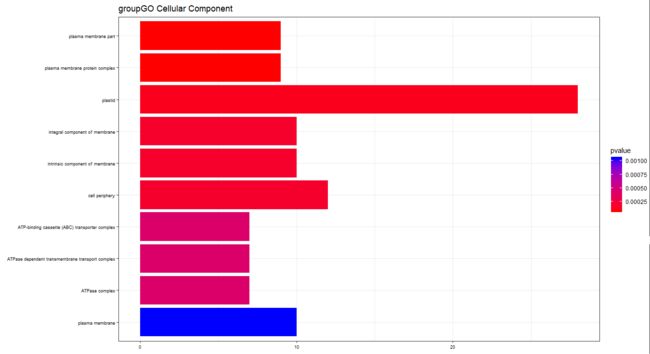

其他一些图
## 热图
## 相关系数图
## 散点图
## heatmap
rm(list=ls()) ## 清除当前环境变量
resdata <- read.csv(file.choose(),header = T , sep = "\t") ## 导入数据PP_data_diffexpr_padj_results
subset(resdata,padj < 0.001 & abs(resdata$log2FoldChange) >= 2) -> diff_gene ## 筛选出差异基因
diff_gene[,8:ncol(diff_gene)] -> heatmap_data ## 提取counts value所在列
## ncol(diff_gene) ## 表示该数据有多少列
rownames(heatmap_data) <- as.array(diff_gene[,1]) ## 添加行名
as.matrix(heatmap_data) -> x ## 转换数据格式
library(pheatmap) # 加载包 如果未安装先install.packages("pheatmap")
pheatmap(x,scale="row",cellwidth=40,cellheight=0.1, ## 设置热图每个格子的宽高
cluster_col = F,cluster_row= F, ## 按行还是按列聚类,一般按行,即基因
main="The PP_data diff_gene heatmap ", (标题)
fontsize=5,treeheight_row = 2,show_rownames= F, ## 是否显示行名(基因名)
cutree_row=1,display_numbers = FALSE, ## 是否显示数值
color = colorRampPalette(c("green","white","red"))(100), ## 设置颜色
clustering_distance_rows = "correlation", #filename="out.pdf"
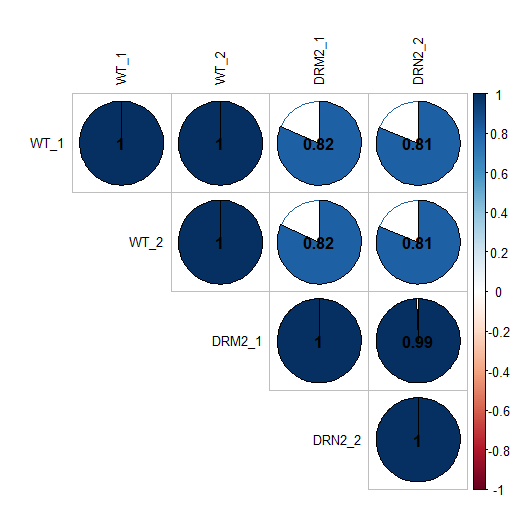
## 相关系数图
## 还是用上面的resdata数据
install.packages("corrplot")
library(corrplot)
resdata[,8:ncol(diff_gene)] -> heatmap_data ## 提取WT以及osdrm2 counts value所在的列
cor(heatmap_data) -> cor_data ## 计算相关系数值
corrplot(cor_data,type = "upper") ## 绘制相关系数图
## counts散点图(也可以用来看相关性)
install.packages("ggplot2")
library(ggplot2)
ggplot(resdata,aes(x= resdata$WT_1,y=resdata$WT_2)) +
geom_point() + xlim(0,80000) + ylim(0,80000) +
geom_abline() + theme_bw() +
xlab("WT_1") +ylab("WT_2") +
theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))
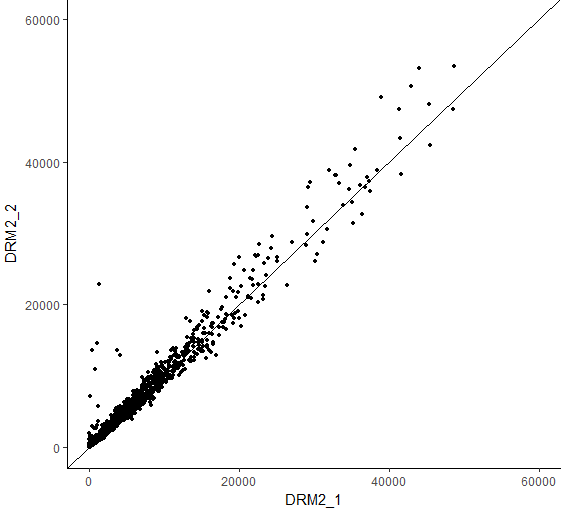
ggplot(resdata,aes(x= resdata$DRM2_1,y=resdata$DRN2_2)) +
geom_point() + xlim(0,80000) + ylim(0,80000) +
geom_abline() + theme_bw() +
xlab("DRM2_1") + ylab("DRM2_2") +
theme(panel.border = element_blank(),panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),axis.line = element_line(colour = "black"))


其他流程
HISAT2 -> HTSeq -> DESEq2
HISAT2 ->StringTie -> Ballgown
kallisto -> sleuth
....
差异分析R包
DESeq / DESeq2
edger
DEGSeq 无重复样本的差异分析
DEXseq
limma
GFOLD ## 无重复样本的差异分析
最后我平时参考的论坛
github 一个开源的网站,大佬们都把源码放置在上面,比如已发表paper的源码
生信技能树
omicshare
rabbit gao个人博客
基因课
小木虫
r-bloggers
感谢爱分享的各位生信人
逛的太多-- 就不一一举例了
一直相信勤能补拙

如有问题请留言。。。