宏基因组的基因定量问题已经困扰我很久啦,今天稍稍梳理了一下,但是还是不大懂,后续分析到底怎么整,什么情况下做基因定量进行差异分析有必要。如果是基因已知数据库的方法,就像MEGAN软件那样,用基因reads count的相对丰度进行物种、功能分析。用自己数据拼接构建的参考基因集,原理还是一样的,但是啊但是参考基因集的物种和功能注释率低。
宏基因组的实验设计思路(我总结的)
-
第一,大范围的微生物资源调查基础性研究,建立参考基因集、基因组集,想要了解微生物的群落结构和功能,像TARA海洋宏基因组项目、人、猪、小鼠这些肠道微生物宏基因组项目;(大样本)
对人、老鼠、猪的肠道微生物建立参考基因集,了解它们肠道微生物群落结构和功能分布,此外结合宿主的生理特征,从而研究年龄、饮食、品种等因素对肠道微生物的影响。人、老鼠、猪的肠道微生物参考基因集的文章都是华大与其他单位合作研究发表的。
海洋微生物建立参考基因集,也是想要了解海洋微生物的群落结构和功能分布,对不同海域进行比较分析。海洋微生物参考基因集的文章是由TARA全球项目组发表的。
解读文章:
宏基因组CNNS文章盘点①——参考基因集
最全的猪肠道菌群参考基因集
-
第二,特殊生态系统,例如藻菌共生系统中,藻和细菌是如何共生代谢的;(单个样本,测得会比较深,重构基因组,一般会结合宏转录组做,想要研究清楚代谢调控机制)
主要使用分箱(Bining:根据contig的GC含量、覆盖度等特征信息对contig进行分组,一般认为分成一个组的contig就是一个微生物基因组了,称genome bin )的方法;
从微生物中重构微生物基因组后,使用必需基因鉴定每个genome bin的完整度和污染度(这里可以自己划定标准,一般完整度90%以上,污染度小于10%),接着对筛选出来的每个genome bin进行物种鉴定(一般就是比对到已知的数据库比对注释,这方面的软件有很多),注释不到门的Bin,可能就是新种了;
后续分析除了物种鉴定、功能注释、代谢通路分析,还有功能基因组的挖掘,通过已知的功能基因(固氮的细菌会有特殊的基因簇),看看哪些有固氮的(当然还有其他功能可以筛选)。
此外就是进行代谢互补分析,看看群落里的微生物是如何合作的。可以看“宏基因组分析揭示微囊藻-细菌的共生关系”,这是一个比较简单的微生物共生群落,分箱得到的Bin个数很少,挑选代谢通路,看看这些细菌在在代谢通路上面是否互补,研究他们之间的共生关系。对于复杂的微生物群落,比如分到100个Bin,将不会从Bin的水平去研究代谢互补了,会根据物种鉴定结果从属、门等比较高的分类水平进行这种研究。在生态环境中比较关注C、N、P、S这些代谢途径。
类似文章:16扩增子结合宏基因组分箱方法进行珊瑚上的一个共生系统:https://www.nature.com/articles/s41598-017-09032-4
-
第三,针对某个因子(时间、空间地理、宿主、理化)设计实验进行宏基因组关联分析,研究该因子对生境中的微生物群落结构和功能的影响;(关联分析也需要一定的样本量,我看人类研究都是上百个样本)
该方法是华大提出来的。
该文章的解读:
http://b4c93a4f.wiz03.com/share/s/2QOjFf14ckoS25rO_m2pfQnt3qbIu82cDk1M2tFSoe3Dt9a4 -
第四,用于不能纯培养的微生物基因组的研究;
像一些微囊藻,它的表面胶鞘会附着一些细菌,很难通过分离纯化把它们分开,但是又想测微囊藻的基因组怎么办?这时就可以用宏基因组学的手段,把他们的DNA都进行测序,都一起拼接,然后分箱,这样在基因组层面,微囊藻和细菌就分开啦~
宏基因组学
"Microbe run the world."
微生物(细菌、病毒、真菌),在我们的生活中无所不在,发挥着不可替代的作用。传统的微生物研究是将它们进行分离纯化,得到单菌株。但是复杂的微生物群落由上千种微生物组成,并且大多数很难通过传统的方法得到分离纯化。在这种情况下,宏基因组学这一项技术就被引入从基因组水平来研究微生物群落。什么是宏基因组学呢?简而言之,就是一个微生物群落的DNA被全部提取出来,接着被随机打断、测序,返回来一大堆短DNA片段。通过宏基因组学技术不仅可以得到微生物群落中的物种结构,还可以进行功能分析,重构难培养微生物的基因组。
以前又慢又贵的Sanger测序,第一批宏基因组测序数据仅有几千条reads。但随着二代高通量技术的诞生,测序成本在不断降低,像人类微生物组计划、地球微生物组计划已经产生了数十亿条reads(万亿的碱基)的数据集了。另外宏基因组技术在生命科学领域的应用越来越广泛了。在医学领域,宏基因组技术用来研究疾病和体内微生物疾病的关联,像二型糖尿病、克罗恩病。在生态领域,宏基因组技术被应用于不同生态系统的微生物群落研究,像牧场土壤、海洋水体、奶牛瘤胃等。在生态毒理学领域,则被用于揭示微生物废水处理中的生物降解机制和了解抗抗生素基因在环境中的分布。
宏基因组学是一种技术,不是一个领域,它只能帮助我们从基因、基因组的层面去了解微生物群落的结构和功能而已。我们需要找到自己的领域,寻找值得研究的问题,设计严谨的实验方案。不要在浩如烟海的数据中,迷失了自己。
上周一位老师来找我们老板咨询,她是实验设计就很好,四个样本代表四种生态型的结皮,每个样本有三个重复,研究沙漠结皮过程中微生物群落的结构、功能的变化,解析结皮系统中的微生物参与的C、N、O、S、H代谢过程。这个生态系统很研究意义,治理沙漠化。
基因定量
这叫啥gene-centirc宏基因组学研究,啥意思,不懂。直接进入正题吧。
基因编码蛋白,发挥它的生物学功能。一般细菌基因组有上千个基因,这就意味着一个微生物群落将会有数百万个基因。在不同的物种中基因可能存在许多变异,但仍然发挥着同样的功能。为了使生物学过程更容易理解,将不同物种有着相似功能的基因分不同蛋白域、基因家族或者同源基因集(像eggNOG、KEGG、TIGRFAM、SEED)。选择基因还是一组基因进行研究要看你的生物学问题了。
在对一个样品中的基因进行定量,需要样品采集、DNA提取、测序这些实验步骤。测序返回一大堆短片短DNA序列(reads),一般长度在75-400bp之间的(取决于所采用的测序技术)。测序并不精准,原始reads经常会存在一些测序误差,例如测错碱基了、插入额外的碱基啦,最高会有1%的错误率。测序数据中会有每个碱基的质量值,能够反映错误的概率,根据质量值进行识别从而去除低质量的reads。接着,比对到已被注释的参考数据库,保留最佳匹配。
参考数据库可以用不同的方法进行构建,可以收集之前被研究过的基因和微生物基因组,也可以直接拼接reads成为更长的片段(预测orf,根据序列相似性原理进行功能注释,这里又要提一下单个基因、基因group的分辨率选择问题了,看自己的需求哈)。每条reads比对到一个数据库某个特定的基因,就表示存在这个基因。通过这种方法,所有的reads能够被“binned”,从而得到每个存在的基因有多少条reads的结果,最后的基因reads的计数数据用来表示每个基因在样品中的丰度。
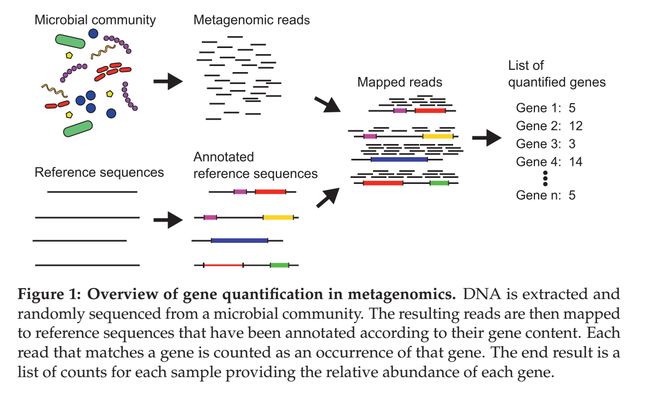
统计学挑战
gene-centic宏基因组重要就是检测基因相对丰度和实验条件的关系,例如不同健康状态人、温度梯度变化等。通过统计学分析根据相对丰度寻找不同群落中的差异基因。
但是宏基因组的gene count数据是离散(因为基因是统计比对到特定基因的reads条数进行定量的)、undersampled的(因为测序深度的问题,不是所有的基因都被测全了,所以会存在用很少的reads用来代表基因,很多基因都是低丰度的),高维(微生物群落的基因多呀,一般会有上千的基因会被鉴定为差异基因,假阳性很高的),存在很高的生物变异(每个样品间的有很多外界影响因素,例如温度、盐度、营养盐水平、PH、宿主的年龄;物种的组成改变群落基因的丰度也会改变,细菌的基因组之间差异很大,像大肠杆菌,核心基因大概有3188个,但是有1500个基因是可以变化的,导致不同菌株之间大概有9万个基因;还有水平基因转移啥的;还有样品中就没有某种微生物,那么它的基因也就没有了,就会产生很多为0的基因count)和技术变异性(样品准备方法不同、测序误差、测序深度不同呀、比对错误呀,但比生物误差要小嘞;另外一个为之技术误差是由数据库引入的,因为现在完整的微生物基因组只有四五千的样子,微生物至少有数千万中吧,这就会导致基因的注释率低),生物重复数很少呀(穷)。
100万的基因,实验组和对照组,即使是5个样本对5个样本,100个样本对100个样本,结果是假阳性非常高,需要非常严格的矫正,矫正到GWAS一样夸张。人有两万个基因,做转录组的时候,有些人为了节约钱,只做3个样本对三个样本实际是不够的,要6个或12个样本以上才能找到真正的差异基因,找到一千个基因,其实是假阳性挺高的。GWAS芯片,人有三十亿个位点,其实只做50万到300万之间,GWAS的P值会卡到很低10-6或10-8。
定表达量不是难事,找差异需要引入新的模型,应该有现成的包来做这件事情。
如果假阳性高,其实并没有什么关系,如果看通路的话,应该没有什么关系。功能单元差异,而不是单个基因的差异。 找出几百万CpG中的哪些在疾病中发生了变化,而这些变化又是如何导致了基因发生了变化,最终导致了人体生病。而做的方法直观上简单的可怕:你有100个癌症病人,100个正常人,每个人身体中都有450K个CpG的位点在测序出来,针对其中的每一个CpG,你都有200个数据对不对?如果这一个CpG在100个正常人中和100个癌症病人中的甲基化水平都差不多,你还会继续怀疑它吗,当然不会!
但如果,你的100个癌症病人普遍在这一个CpG上的甲基化水平高(不太严谨但是很形象地说,就是DNA那个CpG的序列外层越来越多的部分被甲基附集上去了),而那100个普通人的甲基化水平不高,那这个位点就很有嫌疑了对吧。
为什么需要一定量的样本,比如100个?因为如果你只找两个人,一个德国人一个中国人,那个德国人高,那个中国人矮,你能因此就说德国人在人种上比中国人高吗?当然不能……但如果你找到100个德国人,在找到100个中国人,比较以后,就比较可信了,这涉及t.test()里的power的问题。
这就是做研究的艰难:你得找到100个病人,让他们同意治疗,然后耗资几万几十万完成测序,有了数据还要祈祷实验员没有点错试管,数据下来了如果由于年龄、人种等原因,数据差异已经找不到了,你还得想办法修正这些问题,然后你可以开始比较,从几百万位点(基因、SNP、CpG都一样)中找出那些可能有关系的……等你做完这一切,可能一两年已经过去了,而你本科毕业就进入IT界的同学可能已经工资两万多了,这还绝对算是科研中很快节奏的项目了,所以我觉得社会真的应该给科研工作者更多尊重,做最难的活,拿最低的工资。
有时候一把跑出来,几百万位点中,几万甚至十几万都是显著的(做年龄的时候就这样,因为年龄对于人身体的影响太大,可谓是全方位无死角的)!
t.test()里的power的问题。统计学问题
@以上Jimmy师兄说的。
宏基因组的差异基因,自己看着办吧,一个样本的基因会有上百万个,看做人类研究的宏基因组关联分析,样本量都非常大。如果样本量不够,就只做这种表达模式图,我瞎说的?(高、中、低,话说这个界限咋划分呢,用热图是不是就可以啦)。
参考文献
- 海洋微生物参考基因集的建立-Structure and function of the global ocean microbiome-2015
- 人类肠道微生物参考基因集的建立-A human gut microbial gene catalogue establish by metagenomic sequencing-2010
- 人类肠道微生物参考基因组整合-An integrated catalog of reference genes in the human gut microbiome-2014
- 鼠肠道参考基因集-A catalog of the mouse gut metagenome-2015
- 猪肠道微生物参考基因集的建立-A reference gene catalogue of the pig gut microbiome-2016
- 【综述:从宏基因组数据中重构微生物基因组】Recovering complete and draft population genomes from metagenome datasets
- 从海洋沉积物微生物宏基因组中重构98株微生物基因组-Metagenome sequencing and 98 microbial genomes from Juan de Fuca Ridge flank subsurface fluids
- 宏基因组分析揭示微囊藻-细菌的共生关系-Metagenomic Analysis Reveals Symbiotic Relationship among Bacteria in Microcystis-Dominated Community
- 使用TARA项目的微生物宏基因组数据进行基因组重构,根据功能基因挖掘功能微生物-Nitrogen-fixing populations of Planctomycetes and Proteobacteria are abundant in the surface ocean
- 【宏基因组方法学综述-从采样、测序到数据分析】Shotgun metagenomics- from sampling to analysis
- 【综述:宏基因组关联分析】Metagenome-wide association studies fine-mining the microbiome
- 用宏基因组学的手段研究蓝藻基因组A Metagenomic Approach to Cyanobacterial Genomics

如果实在有需要请给我发邮件:[email protected];
也可以关注我的公众号:沈梦圆(PandaBiotrainee)