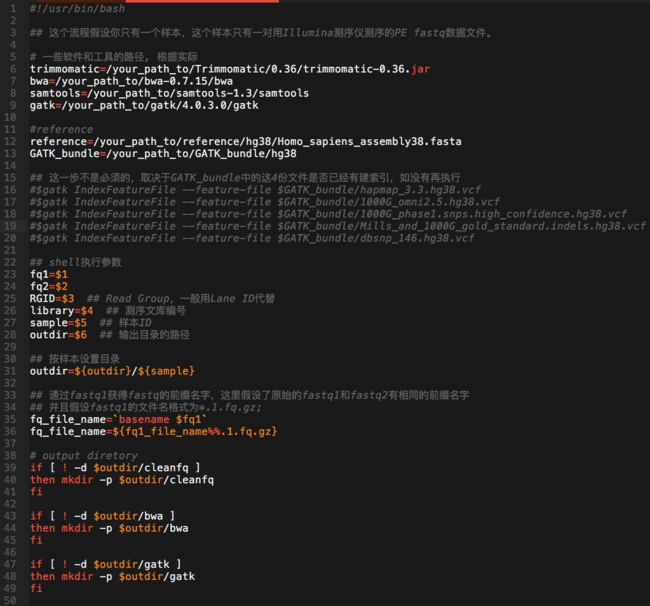
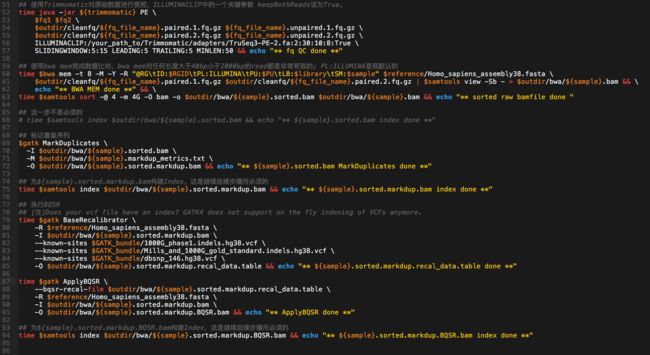
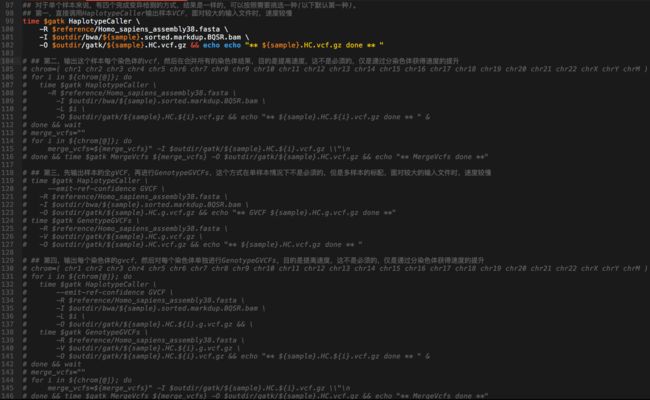
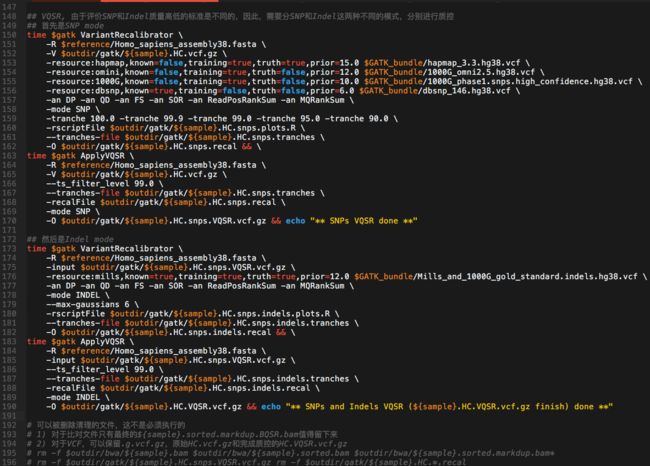
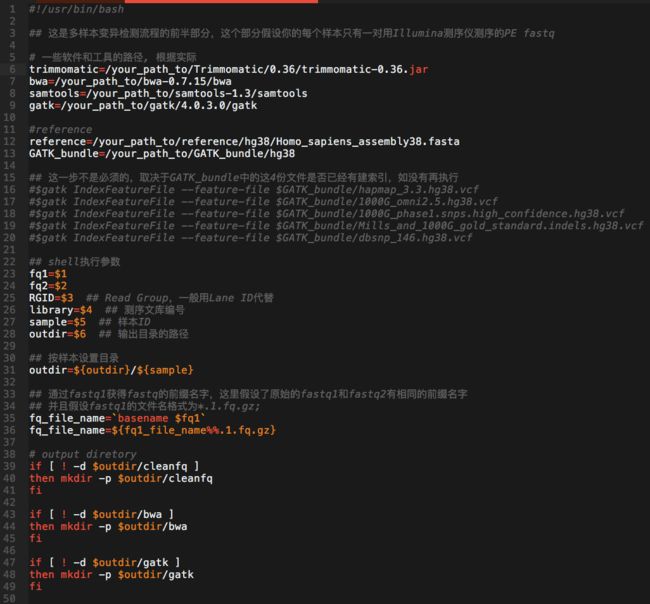
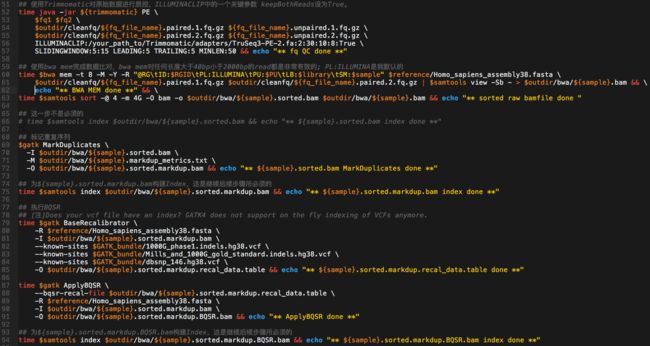
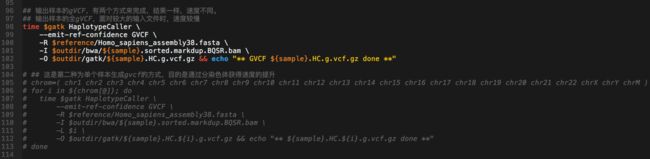
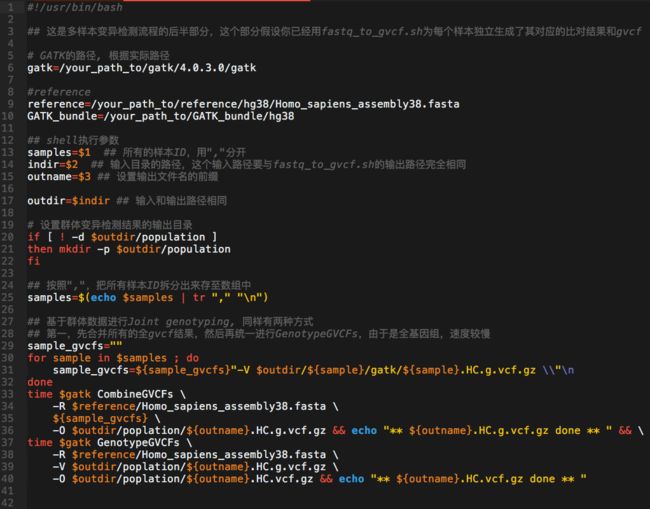
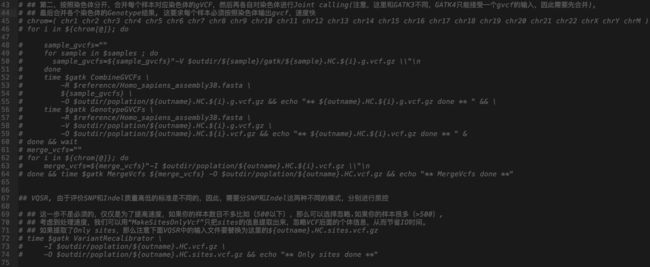
- 7.28日志.王翼
王翼wy
今天到青岛去接妙妙,由于没买上卧铺,昨晚坐了一晚上硬座。到了青岛,妙妙妈带我到了一位女士开的正骨室,对我的身体骨骼进行了系统检查,找到了病根,说不是很严重,只要坚持练习就好康复。这件事让我内心感动,我却从来没这样关心过她。以后要向她学习,多用心关心关心她。我们中午去了一家水饺自助店用餐,吃的很好。下午妙妙妈说去哪儿玩?我看孩子容易迷入视频,就说去游泳吧!(妙妙妈说这两天泳后睡的实发育好)妙妙游了一
- 《独孤残缺》第一百六十一章:还债
卧龙镇吟
“信鸽!”纪茹灵激动的问道:“可是一只全身灰色的鸽子?”李默天点了点头,纪茹灵仿佛受到惊吓一般,往后倒退了几步,除了小灰,没有那个信鸽能够远渡大海来到中原,而且李默天从没有见过小灰这只信鸽,既然他能够说对了鸽子的颜色,就说明他所说的内容也是真实的。其实李默天确实没见过那只信鸽,只是听中田胜一吹嘘过自己有一只了不起的鸽子,所以李默天巧妙的利用了纪茹灵关切亲人安危的心理,在说出信鸽传信这件事后,纪茹灵
- 怎样考研才最高效呢?如何准备呢?
十里li
考研
大学生考研全攻略:备考路径+高效学习法+时间管理考研全流程导航(时间轴)2023-01-012023-02-012023-03-012023-04-012023-05-012023-06-012023-07-012023-08-012023-09-012023-10-012023-11-012023-12-012024-01-012024-02-012024-03-01确定目标院校英语/数学筑基专
- 香奈儿顶级复刻哪家好(高仿香奈儿包价格一览表)
潮奢之家
香奈儿顶级复刻哪家好?这是一个备受关注的话题。香奈儿作为世界顶级奢侈品牌,一直受到众多消费者的喜爱。然而,高昂的价格使得许多消费者望而却步。因此,香奈儿顶级复刻市场应运而生。那么,究竟哪家香奈儿顶级复刻品质更好呢?【重要提醒】文章最下面有联系方式首先,我们需要明确一点,香奈儿品牌对于仿制品的态度是坚决**的。在这里,我们讨论的顶级复刻并非官方渠道产品,而是市面上的仿制品。因此,购买这类产品存在一定
- 4月星座运势——双子座爱社交、天秤人气旺、水瓶座桃花开
筝筝陪你看星星
嗨喽,我是星座博主星芒!4月份的星座运势来啦~本月开始,月运中增加了【健康】部分,希望大家都健康平安,顺心顺意~今天分享的是风象星座们,请同时参考你的上升星座和太阳星座~~太阳或上升双子座整体运势双子座的4月份是社交月,是认识新朋友、开创事业发展的好时机。虽然你可能不愿意抛头露面,但是只要跳出舒适圈,你将有很多收获。另外,可能因为追求外表形象而开销过大,需要提升自己的金钱意识,不要盲目消费。4月6
- 父母要“看得见”孩子
婉君的秘密花园
周星驰在一部电影里有一句这样的精彩台词:“如果你不带金劳(劳士表),不是别人看不起你,而是看不见你。”父母也一样,即使孩子成天在身边,你真的看见过孩子吗?孩子喜欢什么?喜欢哪部电影?喜欢哪个老师?他的好朋友是谁?他的周末或假期喜欢怎么过?如果这些,我们都不知晓,或者不确切,我们怎么能说看得见自己的孩子呢?图片发自App很多时候,作为父母的我们,看得见的是别人家的孩子。你们班谁考试又得一百分,谁比赛
- Linux 压缩、解压文件的 4 种方式。tar、gzip、gunzip、zip、unzip、7z命令使用方法
寒水馨
Linux问题解决方案linux服务器运维压缩解压
Linux压缩、解压文件的4种方式。tar、gzip、gunzip、zip、unzip、7z命令使用方法文章目录Linux压缩、解压文件的4种方式。tar、gzip、gunzip、zip、unzip、7z命令使用方法1.tar1.1.压缩:1.2.解压:1.3.tar命令各参数含义2.gzip、gunzip2.1.压缩:2.2.解压:2.3.gzip、gunzip命令各参数含义2.3.1.gzip
- 很勤劳的人仍然穷,那就要改变思维了
山峰奇美
这个世界有一种人,非常的勤奋,像一个辛勤的小蜜蜂,每天风里来雨里去,但到头来,却依然食不果腹,过着紧巴巴的日子。为什么会这样,他们来不及细想,无暇顾及,因为他们太忙啦!为什会贫穷,应该有以下几个原因吧。没有一个定律,什么都想干。选定一个目标,就要尽可量坚持干下去,人的精力有限,什么都想干什么都要去干,其结果往往是半途而废,很多事情没有干成。便被废弃在了萌芽状态。现代社会,干什么都需要一定的投资,“
- 馒头第六天复盘
向北720
今天非常开心,面条jacky老师在傍晚大约七点半左右就完成了今日的作业提交和打卡任务,然而这个时间我正好在补前三天的行书会阅读任务,一下漏掉了三天的任务,还是非常多的,看到面条的作业,只能先放一放,晚上再点评。到了晚上,确实看到了面条的进步,镜头里jacky老师非常的知性和优雅,明显比之前的视频好很多,朗读感悟部分内容的结构也是非常的清晰了,美中不足的是最后的升华部分可以再高一点。给出点评后,面条
- #ifdef和#if 的应用差异
woainizhongguo.
C/C++单片机
在嵌入式软件开发中,#ifdef和#if都是预处理指令,用于条件编译,但它们的应用场景和逻辑有显著差异:一、ifdefifndef用法1.#ifdef/#ifndef作用:检查某个宏是否被定义(无论其值如何)。语法:#ifdefMACRO_NAME//如果MACRO_NAME被定义,编译此部分代码#endif#ifndefMACRO_NAME//如果MACRO_NAME未被定义,编译此部分代码#e
- 你想成为什么的人,就得做什么样的事
大道行者_
你发现没有,生活中充满负能量的那些人,抱怨的东西几乎是不变的。跟他们聊天,聊不出任何新鲜的东西。要么先吹嘘自己的经历,吹嘘自己去过的地方,可那些事他自己已经提了无数遍,早已听到耳朵出茧;要么抱怨,抱怨工作,抱怨付出没有回报,抱怨别人只是运气好。永远在放大自己的付出,责怪整个世界没眼光。怎么说呢,如果我们每个人都是一个半圆,人人都有自己的半径。有的人拼命充实自己,把半径越拉越长,自然面积越来越大;可
- 我见即我思第824天
若如初梘
我见:悲观者在每个机会中,看到的都是困难;而乐观者则在困境中洞察良机。——(by温斯顿·邱吉尔)(摘自句读中语录)我思:机会都是给有准备的人,并不是说机会摆在你面前,你就可以获得,是需要我买来进行一定的操作,才能获得成功的。所以在那些悲观这样的机会,大部分都是困难。而在乐观的眼里,所有的困难又何尝不是一种机会,只要我们能抓住,就能一飞冲天。我们要用乐观的心态看待世间的万物,只有这样,我们才有足够的
- 郁闷的一下午
方丽华
疲惫不堪的一周,在今晚和同事们的一通发泄之后,心渐渐平静,睡得太晚,每天感觉昏昏沉沉的,打开,总算有块属于自己的安静之地。刘同学又偷别人东西了,别人的东西,拿来,改成自己的名字,我寻问,她眼神里竟没有丝毫的愧疚,理所应当的样子,教育了不知多少次,道理她都懂,就是不改。文明学生的评选,影响不了她,小组之间的竞争,她无所谓,感觉自己真失败。
- 【华为419机考真题】服务器能耗统计,JAVA 题解
梦想橡皮擦
华为服务器java华为OD机试华为OD
最近更新的博客华为od2023|什么是华为od,od薪资待遇,od机试题清单华为OD机试真题大全,用Python解华为机试题|机试宝典【华为OD机试】全流程解析+经验分享,题型分享,防作弊指南华为od机试,独家整理已参加机试人员的实战技巧本篇题解:服务器耗能题目描述服务器有三种运行状态:空载,单任务,多任务,每个时间片的能耗的分别为111、333、444,每个任务由起始时间片和结束时间片定义运行时
- LVS调度算法+防火墙解决轮询调度问题+会话解决
甜辣小悦羊
lvs服务器运维
lvs的调度算法类型分配:依据负载状态静态方法:仅根据算法本身进行调度,不考虑RS的负载情况动态方法:主要根据每RS当前的负载状态及调度算法进行调度Overhead=value较小的RS将被调度静态调度方法:RR(roundrobin):轮询RS分别被调度,当RS配置有差别时不推荐WRR(WeightedRR):加权轮询根据RS的配置进行加权调度,性能差的RS被调度的次数少SH(SourceHas
- LangChain4j入门:Java开发者的AI应用开发指南
半夜偷你家裤衩子
LangChain4jjava人工智能开发语言LangChain4j
在AI浪潮席卷全球的今天,Java开发者如何快速上手大语言模型应用开发?LangChain4j为我们提供了完美的解决方案!前言:为什么Java开发者需要LangChain4j?想象一下,你正在开发一个企业级应用,需要集成ChatGPT来提供智能客服功能。传统方式需要直接调用OpenAIAPI,处理复杂的HTTP请求、错误重试、上下文管理等问题。而使用LangChain4j,几行代码就能搞定:Cha
- LVS的10种调度算法
蜡笔晓心
其他
1.1静态算法:1.1.1rr(roundrobin):轮询调度算法:轮询调度算法的原理就是依次将用户的访问请求,平均的分配到每一台web服务节点上,从1开始,到最后一台服务器节点结束,然后在开始新一轮的循环,这种算法简单,但是没有考虑到每台节点服务器的具体性能1.1.2wrr(weight):权重调度算法由于每台服务器的性能会高低不同,wrr将会根据管理员设定的权重值来分配访问请求,权重值越大的
- 算法工程师必备:数据结构10大经典算法详解
数据结构与算法学习
数据结构与算法宝典算法数据结构ai
算法工程师必备:数据结构10大经典算法详解关键词:数据结构、经典算法、时间复杂度、应用场景、代码实现摘要:本文是算法工程师的“算法工具箱”指南,系统讲解数据结构领域最核心的10大经典算法(快速排序、归并排序、二分查找、深度优先搜索DFS、广度优先搜索BFS、动态规划、贪心算法、KMP字符串匹配、哈希算法、并查集)。通过生活案例、代码示例、复杂度分析和实战场景,帮你彻底掌握这些算法的原理与应用,真正
- 时间有限,分清事情的轻重缓急
雁南征
开始进入繁忙的季节了,白天工作上很多事情要处理。余下业余时间有限,待处理的事情太多,应接不暇。最近待完成的文章太多,而时间又总是太少,一天仅有24小时,除工作和睡觉吃饭,好象所剩无几,前段时间还有时间来看电视剧,这一两周已经停看了。健身、写作、阅读,都是需要时间填充的,所以得分清主次来。首先是健身,身体是第一位的,散步、跑步、健身、八段锦、俯卧撑、平板支撑、打坐冥想,每天安排的这些事情应该优先完成
- 古驰高仿衣服在哪买,试试这10个渠道
金源皮具
古驰高仿衣服在哪买,试试这10个渠道在时尚界,古驰(Gucci)无疑是一个响当当的名字,其独特的设计和高昂的价格让许多时尚爱好者望而却步。然而,对于那些渴望拥有古驰风格但又预算有限的人来说,高仿古驰衣服成为了一个不错的选择。本文将为您揭秘10个购买高仿古驰衣服的渠道,让您在不破费的情况下也能享受奢华的时尚体验。1.电商平台:淘宝、京东、拼多多等国内大型电商平台上有许多卖家提供高仿古驰衣服。这些平台
- 访问容器中的元素
tal0n
上一篇遗留的问题在上一篇中我们实现了一个类似内建数组的容器,但是这个容器包含了内建数组的缺陷由于operator[]返回的类型T&导致用户可以获取到容器内部元素的地址,在容器不存在以后这个指针依然存在。由于维护了容器到数据的指针关系,我们过多的暴漏了容器的内部机制。用户可以使用指针直接访问容器内部,一旦容器内部占用的内存发生变化,将导致用户错误。导致resize这类的函数很难实现。模拟指针c++中
- 星座占星三王星深层次意义
亮天机
image每一个地球人的命盘上都有天王、海王与冥王。这三颗星代表了人类最极端的三种潜在趋势,即——神性、魔性与变异。由于他们的存在,所以任何一个人都有产生任何一种变化的可能性。所以再善良的人都堕落的空隙、再邪恶的人都有成佛的可能,再顺从的人被压迫久了都会叛变。image天王——蛰伏在人类基因中的不确定因子。我们生活的领域里充满各式各样的惯性。物体有惯性,总试图维持当前的运动状态;人有惯性,总拒绝任
- YXG362~037抓复习的方法
丁妞森娃
没有一种知识是不需要复习巩固的,没有一种能力是不需要反复训练的。第一条日常进行知识性复习第二课时新授前要复习第一课时的内容,复习朗读、词、多音字、复习概括、分段。第二篇课文的第一课时先复习上一篇课文的要点。一个单元上两个星期,上星期学的内容到下一星期也要复习,一周一次基本知识的过关要形成惯例,小步子的小复习走得扎实,大步子的复,中后等学生跟不上。第二条单元进行整理性复习。一个单元结束后,复习主要依
- 听听自己的声音
绿色番茄
刚准备洗漱,手机发来提示音,原来是方老师提交了作业忽然想起今天某人第一次提交作业,抓耳挠腮,像憋着大的……最近几天其实都有在写,只是每每满腹的话,却不能一吐为快,为啥呢,写了一半,实在写不下去的有三篇,哎呀,写着写着,就觉得没有了写的必要思来想去,我分析,应该是我自愈能力太强了,上一秒被hans气得要吐血,发誓要好好写一篇,记录他的恶行,下一秒心底又有了一个声音在说:亲生的…亲生的…是不是我的方式
- 微信小程序入门实例_____从零开始 开发一个“旅行清单 ”微信小程序
数码小沙
微信小程序微信小程序小程序
前面的博文中。我们陆续学习与开发了记账等一些实用实用小程序的开发过程,今天来打造一个适合出行场景的工具——“旅行清单小程序”。无论是短途游玩还是长途旅行,它都能帮你梳理需要携带的物品,避免遗漏。下面就跟着步骤,一步步实现这个小程序。再次体验开发者的快乐一、开发小程序员前的准备工作1.工具检查确保微信开发者工具已安装并更新到最新版本。若未安装,打开微信公众平台(微信公众平台),在页面底部找到“下载”
- lvs调度算法(10种)
beyoundout
lvs算法
一、静态算法(不考虑后端真实服务器的负载情况,按算法该谁就分配给谁)1.rr(RoundRobin)轮询算法算法原理:将外部请求按顺序轮流分配到集群中的真实服务器上,它均等地对待每一台服务器,而不管服务器上实际的连接数和系统负载举例:就像在食堂打饭,有三个打饭窗口。学生们排成一队从餐厅门口进入食堂,依次到第一个窗口、第二个窗口、第三个窗口打饭,后面的学生再从第一个窗口循环,每个窗口平等地接待学生,
- 计算机网络中:传输层和网络层之间是如何配合的
woainizhongguo.
计算机网络计算机网络网络
可以把网络层和传输层想成一个“快递系统”:网络层(IP层)=邮政系统:只负责把“包裹”(IP数据报)从A地搬到B地,不保证顺序、不保证不丢、不保证不重复。传输层(TCP/UDP层)=快递公司:在邮政系统之上提供“增值服务”——对TCP来说,就是可靠、按序、不重复、流量受控的端到端运输;对UDP来说,就是“无连接、尽力而为”的快送。下面用一次典型的TCP通信说明两者如何“接力配合”。1.建立连接(三
- 华为服务器管理工具(Intelligent Platform Management Interface)
小小玫瑰大智慧
华为服务器运维
一、核心功能与技术架构硬件级监控与控制全维度传感器管理:实时监测CPU、内存、硬盘、风扇、电源等硬件组件的温度、电压、转速等参数,支持超过200种传感器类型。例如,通过IPMI命令ipmitoolsdrelist可快速获取服务器传感器状态,并通过正则表达式提取关键指标。远程操作能力:支持远程开关机、重启、BIOS设置调整、固件升级等操作,即使服务器操作系统崩溃或网络中断,仍可通过独立BMC芯片实现
- 拼多多官方返利新动向,高省App引领购物省钱新趋势
古楼
电商行业的快速发展带来了无数的新趋势和新机遇,而拼多多官方返利的新趋势无疑是其中的一大亮点。高省App作为这一趋势的敏锐洞察者和积极参与者,致力于帮助用户精准把握这些新机遇。通过高省App,用户可以及时了解拼多多官方返利的最新政策和活动信息,从而做出更加明智的购物决策。同时,高省App还提供了专业的数据分析工具,帮助用户分析自己的消费行为和省钱效果,让省钱之路更加清晰和明确。我们在开始讲今天的文章
- 在家有哪些能做的赚钱项目?在家挣钱的兼职有哪些?
古楼
高省app是浙江的一家专业的网购省钱APP,致力于为用户提供更好的网购优惠,实现购物优惠最大化。高省是由杭州长孚科技有限公司开发的一款专门帮助淘宝天猫卖家、品牌代运营等商家省钱的app,也是目前国内唯一一款能让消费者真正省钱的APP,高省为所有用户提供“分享赚佣金”和“邀请他人赚佣金”两种赚钱模式。在购物、旅游、学习中用到优惠券的时候,可以在高省APP上领取哦。一、自用省钱自用省钱是指用户在购买产
- java责任链模式
3213213333332132
java责任链模式村民告县长
责任链模式,通常就是一个请求从最低级开始往上层层的请求,当在某一层满足条件时,请求将被处理,当请求到最高层仍未满足时,则请求不会被处理。
就是一个请求在这个链条的责任范围内,会被相应的处理,如果超出链条的责任范围外,请求不会被相应的处理。
下面代码模拟这样的效果:
创建一个政府抽象类,方便所有的具体政府部门继承它。
package 责任链模式;
/**
*
- linux、mysql、nginx、tomcat 性能参数优化
ronin47
一、linux 系统内核参数
/etc/sysctl.conf文件常用参数 net.core.netdev_max_backlog = 32768 #允许送到队列的数据包的最大数目
net.core.rmem_max = 8388608 #SOCKET读缓存区大小
net.core.wmem_max = 8388608 #SOCKET写缓存区大
- php命令行界面
dcj3sjt126com
PHPcli
常用选项
php -v
php -i PHP安装的有关信息
php -h 访问帮助文件
php -m 列出编译到当前PHP安装的所有模块
执行一段代码
php -r 'echo "hello, world!";'
php -r 'echo "Hello, World!\n";'
php -r '$ts = filemtime("
- Filter&Session
171815164
session
Filter
HttpServletRequest requ = (HttpServletRequest) req;
HttpSession session = requ.getSession();
if (session.getAttribute("admin") == null) {
PrintWriter out = res.ge
- 连接池与Spring,Hibernate结合
g21121
Hibernate
前几篇关于Java连接池的介绍都是基于Java应用的,而我们常用的场景是与Spring和ORM框架结合,下面就利用实例学习一下这方面的配置。
1.下载相关内容: &nb
- [简单]mybatis判断数字类型
53873039oycg
mybatis
昨天同事反馈mybatis保存不了int类型的属性,一直报错,错误信息如下:
Caused by: java.lang.NumberFormatException: For input string: "null"
at sun.mis
- 项目启动时或者启动后ava.lang.OutOfMemoryError: PermGen space
程序员是怎么炼成的
eclipsejvmtomcatcatalina.sheclipse.ini
在启动比较大的项目时,因为存在大量的jsp页面,所以在编译的时候会生成很多的.class文件,.class文件是都会被加载到jvm的方法区中,如果要加载的class文件很多,就会出现方法区溢出异常 java.lang.OutOfMemoryError: PermGen space.
解决办法是点击eclipse里的tomcat,在
- 我的crm小结
aijuans
crm
各种原因吧,crm今天才完了。主要是接触了几个新技术:
Struts2、poi、ibatis这几个都是以前的项目中用过的。
Jsf、tapestry是这次新接触的,都是界面层的框架,用起来也不难。思路和struts不太一样,传说比较简单方便。不过个人感觉还是struts用着顺手啊,当然springmvc也很顺手,不知道是因为习惯还是什么。jsf和tapestry应用的时候需要知道他们的标签、主
- spring里配置使用hibernate的二级缓存几步
antonyup_2006
javaspringHibernatexmlcache
.在spring的配置文件中 applicationContent.xml,hibernate部分加入
xml 代码
<prop key="hibernate.cache.provider_class">org.hibernate.cache.EhCacheProvider</prop>
<prop key="hi
- JAVA基础面试题
百合不是茶
抽象实现接口String类接口继承抽象类继承实体类自定义异常
/* * 栈(stack):主要保存基本类型(或者叫内置类型)(char、byte、short、 *int、long、 float、double、boolean)和对象的引用,数据可以共享,速度仅次于 * 寄存器(register),快于堆。堆(heap):用于存储对象。 */ &
- 让sqlmap文件 "继承" 起来
bijian1013
javaibatissqlmap
多个项目中使用ibatis , 和数据库表对应的 sqlmap文件(增删改查等基本语句),dao, pojo 都是由工具自动生成的, 现在将这些自动生成的文件放在一个单独的工程中,其它项目工程中通过jar包来引用 ,并通过"继承"为基础的sqlmap文件,dao,pojo 添加新的方法来满足项
- 精通Oracle10编程SQL(13)开发触发器
bijian1013
oracle数据库plsql
/*
*开发触发器
*/
--得到日期是周几
select to_char(sysdate+4,'DY','nls_date_language=AMERICAN') from dual;
select to_char(sysdate,'DY','nls_date_language=AMERICAN') from dual;
--建立BEFORE语句触发器
CREATE O
- 【EhCache三】EhCache查询
bit1129
ehcache
本文介绍EhCache查询缓存中数据,EhCache提供了类似Hibernate的查询API,可以按照给定的条件进行查询。
要对EhCache进行查询,需要在ehcache.xml中设定要查询的属性
数据准备
@Before
public void setUp() {
//加载EhCache配置文件
Inpu
- CXF框架入门实例
白糖_
springWeb框架webserviceservlet
CXF是apache旗下的开源框架,由Celtix + XFire这两门经典的框架合成,是一套非常流行的web service框架。
它提供了JAX-WS的全面支持,并且可以根据实际项目的需要,采用代码优先(Code First)或者 WSDL 优先(WSDL First)来轻松地实现 Web Services 的发布和使用,同时它能与spring进行完美结合。
在apache cxf官网提供
- angular.equals
boyitech
AngularJSAngularJS APIAnguarJS 中文APIangular.equals
angular.equals
描述:
比较两个值或者两个对象是不是 相等。还支持值的类型,正则表达式和数组的比较。 两个值或对象被认为是 相等的前提条件是以下的情况至少能满足一项:
两个值或者对象能通过=== (恒等) 的比较
两个值或者对象是同样类型,并且他们的属性都能通过angular
- java-腾讯暑期实习生-输入一个数组A[1,2,...n],求输入B,使得数组B中的第i个数字B[i]=A[0]*A[1]*...*A[i-1]*A[i+1]
bylijinnan
java
这道题的具体思路请参看 何海涛的微博:http://weibo.com/zhedahht
import java.math.BigInteger;
import java.util.Arrays;
public class CreateBFromATencent {
/**
* 题目:输入一个数组A[1,2,...n],求输入B,使得数组B中的第i个数字B[i]=A
- FastDFS 的安装和配置 修订版
Chen.H
linuxfastDFS分布式文件系统
FastDFS Home:http://code.google.com/p/fastdfs/
1. 安装
http://code.google.com/p/fastdfs/wiki/Setup http://hi.baidu.com/leolance/blog/item/3c273327978ae55f93580703.html
安装libevent (对libevent的版本要求为1.4.
- [强人工智能]拓扑扫描与自适应构造器
comsci
人工智能
当我们面对一个有限拓扑网络的时候,在对已知的拓扑结构进行分析之后,发现在连通点之后,还存在若干个子网络,且这些网络的结构是未知的,数据库中并未存在这些网络的拓扑结构数据....这个时候,我们该怎么办呢?
那么,现在我们必须设计新的模块和代码包来处理上面的问题
- oracle merge into的用法
daizj
oraclesqlmerget into
Oracle中merge into的使用
http://blog.csdn.net/yuzhic/article/details/1896878
http://blog.csdn.net/macle2010/article/details/5980965
该命令使用一条语句从一个或者多个数据源中完成对表的更新和插入数据. ORACLE 9i 中,使用此命令必须同时指定UPDATE 和INSE
- 不适合使用Hadoop的场景
datamachine
hadoop
转自:http://dev.yesky.com/296/35381296.shtml。
Hadoop通常被认定是能够帮助你解决所有问题的唯一方案。 当人们提到“大数据”或是“数据分析”等相关问题的时候,会听到脱口而出的回答:Hadoop! 实际上Hadoop被设计和建造出来,是用来解决一系列特定问题的。对某些问题来说,Hadoop至多算是一个不好的选择,对另一些问题来说,选择Ha
- YII findAll的用法
dcj3sjt126com
yii
看文档比较糊涂,其实挺简单的:
$predictions=Prediction::model()->findAll("uid=:uid",array(":uid"=>10));
第一个参数是选择条件:”uid=10″。其中:uid是一个占位符,在后面的array(“:uid”=>10)对齐进行了赋值;
更完善的查询需要
- vim 常用 NERDTree 快捷键
dcj3sjt126com
vim
下面给大家整理了一些vim NERDTree的常用快捷键了,这里几乎包括了所有的快捷键了,希望文章对各位会带来帮助。
切换工作台和目录
ctrl + w + h 光标 focus 左侧树形目录ctrl + w + l 光标 focus 右侧文件显示窗口ctrl + w + w 光标自动在左右侧窗口切换ctrl + w + r 移动当前窗口的布局位置
o 在已有窗口中打开文件、目录或书签,并跳
- Java把目录下的文件打印出来
蕃薯耀
列出目录下的文件文件夹下面的文件目录下的文件
Java把目录下的文件打印出来
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2015年7月11日 11:02:
- linux远程桌面----VNCServer与rdesktop
hanqunfeng
Desktop
windows远程桌面到linux,需要在linux上安装vncserver,并开启vnc服务,同时需要在windows下使用vnc-viewer访问Linux。vncserver同时支持linux远程桌面到linux。
linux远程桌面到windows,需要在linux上安装rdesktop,同时开启windows的远程桌面访问。
下面分别介绍,以windo
- guava中的join和split功能
jackyrong
java
guava库中,包含了很好的join和split的功能,例子如下:
1) 将LIST转换为使用字符串连接的字符串
List<String> names = Lists.newArrayList("John", "Jane", "Adam", "Tom");
- Web开发技术十年发展历程
lampcy
androidWeb浏览器html5
回顾web开发技术这十年发展历程:
Ajax
03年的时候我上六年级,那时候网吧刚在小县城的角落萌生。传奇,大话西游第一代网游一时风靡。我抱着试一试的心态给了网吧老板两块钱想申请个号玩玩,然后接下来的一个小时我一直在,注,册,账,号。
彼时网吧用的512k的带宽,注册的时候,填了一堆信息,提交,页面跳转,嘣,”您填写的信息有误,请重填”。然后跳转回注册页面,以此循环。我现在时常想,如果当时a
- 架构师之mima-----------------mina的非NIO控制IOBuffer(说得比较好)
nannan408
buffer
1.前言。
如题。
2.代码。
IoService
IoService是一个接口,有两种实现:IoAcceptor和IoConnector;其中IoAcceptor是针对Server端的实现,IoConnector是针对Client端的实现;IoService的职责包括:
1、监听器管理
2、IoHandler
3、IoSession
- ORA-00054:resource busy and acquire with NOWAIT specified
Everyday都不同
oraclesessionLock
[Oracle]
今天对一个数据量很大的表进行操作时,出现如题所示的异常。此时表明数据库的事务处于“忙”的状态,而且被lock了,所以必须先关闭占用的session。
step1,查看被lock的session:
select t2.username, t2.sid, t2.serial#, t2.logon_time
from v$locked_obj
- javascript学习笔记
tntxia
JavaScript
javascript里面有6种基本类型的值:number、string、boolean、object、function和undefined。number:就是数字值,包括整数、小数、NaN、正负无穷。string:字符串类型、单双引号引起来的内容。boolean:true、false object:表示所有的javascript对象,不用多说function:我们熟悉的方法,也就是
- Java enum的用法详解
xieke90
enum枚举
Java中枚举实现的分析:
示例:
public static enum SEVERITY{
INFO,WARN,ERROR
}
enum很像特殊的class,实际上enum声明定义的类型就是一个类。 而这些类都是类库中Enum类的子类 (java.l