chip-seq
参考的资料-三个链接
总结目录 一不小心就把ChIP-seq数据分析教程给写完了
一个系列第10篇:ATAC-Seq、ChIP-Seq、RNA-Seq整合分析(本系列完结,内附目录)
给学徒ChIP-seq数据处理流程
最最基本概念
ChIP实验(Chromatin immunoprecipitation)即染色质免疫沉淀,根据DNA与蛋白质相互作用的原理,分离富集与感兴趣的蛋白相互作用的DNA。ChIP-Seq即对分离得到的DNA扩增测序,然后通过分析得到DNA的富集区域也称为peaks,同时可以鉴定过表达的序列motif以及进行功能注释分析。
-
ATAC-seq(Assay for Transposase-Accessible Chromatin with high throughput sequencing) 是2013年由斯坦福大学William J. Greenleaf和Howard Y. Chang实验室开发的用于研究染色质可及性(通常也理解为染色质的开放性)的方法, 原理是通过转座酶Tn5容易结合在开放染色质的特性,然后对Tn5酶捕获到的DNA序列进行测序。
真核生物的核DNA并不是裸露的,而是与组蛋白结合形成染色体的基本结构单位核小体,核小体再经逐步的压缩折叠最终形成染色体高级结构(如人的DNA链完整展开约2m长,经过这样的折叠就变成了纳米级至微米级的染色质结构而可以储存在小小的细胞核)。而DNA的复制转录是需要将DNA的紧密结构打开,从而允许一些调控因子结合(转录因子或其他调控因子)。这部分打开的染色质,就叫开放染色质,打开的染色质允许其他调控因子结合的特性称为==染色质的可及性==(chromatin accessibility)。因此,认为染色质的可及性与转录调控密切相关。
开放染色质的研究方法有ATAC-seq以及传统的DNase-Seq及FAIRE-seq等,ATAC-Seq由于所需细胞量少,实验简单,可以在全基因组范围内检测染色质的开放状态,目前已经成为研究染色质开放性的首选技术方法。==ATAC-Seq与ChIP-Seq的不同的是==ATAC-Seq是全基因组范围内检测染色质的开放程度,可以得到全基因组范围内的蛋白质可能结合的位点信息,一般用于不知道特定的转录因子,用此方法与其他方法结合筛查感兴趣的特定调控因子;但是ChIP-Seq是明确知道感兴趣的转录因子是什么,根据感兴趣的转录因子设计抗体去做ChIP实验拉DNA,验证感兴趣的转录因子是否与DNA存在相互作用。ATAC-Seq、ChIP-Seq、Dnase-Seq、MNase-Seq、FAIRE-Seq整体的分析思路一致,找到富集区域,对富集区域进行功能分析。
ChIP-Seq是揭示特定转录因子或蛋白复合物的结合区域,实际是研究DNA和蛋白质的相互作用,利用抗体将蛋白质和DNA一起富集,并对富集到的DNA进行测序。
DNase-Seq、ATAC-Seq、FAIRE-Seq都是用来研究开放染色质区域。DNase-Seq是用的DNase I内切酶识别开放染色质区域,而ATAC-seq是用的==Tn5==转座酶,随后进行富集和扩增;FAIRE-Seq是先进行超声裂解,然后用酚-氯仿富集。
MNase-Seq是用来鉴定核小体区域。
文库构建包括以下5步骤:
蛋白质与DNA的交联
超声打断DNA链
加附有抗体的磁珠用于免疫沉淀
解交联,纯化DNA
DNA片段大小选择和PCR扩增
富集到的DNA片段只有一部分是真实的信号(感兴趣的蛋白结合的DNA区域),这个比例取决于number of active binding sites, the number of starting genomes, and the efficiency of the IP.
-
ChIP-Seq富集序列存在以下特点:
- 开放染色质区域比紧密区域更易打断;
- 重复序列会出现似乎被富集的现象
- 序列在整个基因组上不均匀分布
-
bed文件解释
- https://vip.biotrainee.com/d/167-bed
老大提出一些疑问
http://www.biotrainee.com/thread-845-1-1.html
现罗列出来如下,边学习边思考
人老了,容易忘事,而且事情也实在是太多了!
本来计划解决的表观的18个问题,一直没抽出空来,而且时间久远了,全部忘记了。找了好久才从微信聊天记录找到。还是放在这个论坛吧,反正我很容易查找我的论坛的帖子!
1、 所有的蛋白都能被修饰吗?都能被甲基化,乙基化,磷酸化等等?http://www.biotrainee.com/thread-450-1-1.html
2、所有的蛋白都能找到抗体吗?http://www.biotrainee.com/thread-451-1-1.html 抗体有效性如何评价?蛋白质有抗体,蛋白质被修饰后修饰位点又有特异性抗体?抗体获取实验环节耗时如何?
3、蛋白质的修饰,主要就是甲基化,乙酰化和磷酸化! 这3种修饰(methyltranferase/acethytransferase/phosphatase),有一些是蛋白质特异性,比如只针对histone,有一些是氨基酸特异性,有一些是广谱的。 甲基化的重点是arginine和lysine,包括PRMT系列和KMT系列,去甲基化主要有JMJD6和KMD系列。 其中PRMT4改名为CARM1。 KMT6改名为EZH2 。 乙酰化的重点是 HATs家族 ,去乙酰化是 HDACs。虽然它们的名字里面带有histone,但其实它们可以作用于其它蛋白质。 磷酸化只能发生在Serine/Threonine/Tyrosine这3个氨基酸上面,统称为kinase,主要是MAPK系列,去磷酸化由有DUSP系列,PTP系列,PPP系列基因。 这个重点是酶与底物的结合!
4、那么既然这么多修饰,我们随便拿其中一个修饰来看看,比如HAT酶,就是给组蛋白的K氨基酸乙酰化的酶,迄今为止,已鉴定的人HAT可分为以下五大类。 (1)Gcn5相关N—乙酰基转移酶(GNAT)超家族: (2)P300/CBP: (3)TAF250: (4)核受体共激活因子: (5)MYST家族: 这是第一个协作任务,那么能把这个完整列表得到吗? gene symbol即可 ,加上分类
5、上面我们说到,组蛋白的K氨基酸乙酰化的酶叫HAT系列,你们可以在genecard上面查一下,就明白了,但实际上,它们应该改名叫做KAT,因为它的重点是K(氨基酸),不是H(histone),这个你们记住,第二个协作任务,把里面的信息搞清楚,历史遗留原因,它被这样命名了。
6、既然组蛋白的K氨基酸可以被乙酰化,根据动态平衡的定律,必然有去乙酰化的酶。 组蛋白去乙酰化酶(histone deacetylase,HDAC)是一类蛋白酶,对染色体的结构修饰和基因表达调控发挥着重要的作用。一般情况下,组蛋白的乙酰化有利于DNA与组蛋白八聚体的解离,核小体结构松弛,从而使各种转录因子和协同转录因子能与DNA结合位点特异性结合,激活基因的转录。而组蛋白的去乙酰化则发挥相反的作用。 而人有18个HDACs,分成4类! 并不是所有的HDACs都被做过CHIP-seq,你们帮我备忘一下,我需要查一查哪一些做过。第三个协作任务。那么能把这个HDACs完整列表得到吗? gene symbol即可 ,加上分类
7、谈到CHIP-seq这个技术,它只是表观研究领域最早最成熟的而已,其实目前有非常多的类似的技术,大家在我们-生信技能树论坛的表观版块可以找到。http://www.biotrainee.com/forum-47-1.html 这是我们的第4个协作任务,你们需要熟悉这个版块,把还没有介绍过的seq技术补全,搜集资料补全即可。
8、前面谈到乙酰化和去乙酰化,初学者会有一个绝对的观点,就是乙酰化就是促进转录,去乙酰化就是抑制转录。同理,很多人会以为,H3K4, H3K36, and H3K79, 这样的active marker,就是激活转录,H3K9, H3K27, and H4K20.这样的repression marker就是抑制转录。同理,大家做一个总结,大家公认的这样的marker的表格,这个是第5个协作任务。大家领取协作任务之前,在这个聊天小群聊一下。
9、前面我们提到了H3K4, H3K36, and H3K79, ,H3K9, H3K27, and H4K20 这些东西,大家可能有人都还不知道,这些组蛋白修饰是什么意思,强烈建议看一下一篇综述,了解组蛋白(包括其它蛋白)可以被哪些修饰,有哪些被研究的修饰可能性。Chromatin Modifications and Their Function
10、接下来,我们说一说转录因子,真核生物的转录非常复杂,转录因子和转录复合物扮演的角色非常重要。转录因子,必然是蛋白质,那么比如也是人体的基因编码的。人人类的基因是已知的,就那么两万多个。而转录因子的定义是已知的,所以理论上我们是可以拿到所有的转录因子列表的,对人类来说,参考:http://www.biotrainee.com/thread-131-1-1.html。而且可以根据重要性,对转录因子进行排序。这是第6个协作任务。转录因子的总结
11、谈到了转录因子和组蛋白,那么我们就要思考一个问题。它们是最主要的CHIP-seq的IP,那么是不是只有转录因子和组蛋白可以做CHIP-seq呢?显然不是,转录复合物,POLY II 的各个组分都可以,PRC1,PRC2也可以,大家还可以提出其它的复合物。这是第7个协作任务,做一个完善的转录相关复合物列表。
12、既然我们把转录因子和组蛋白,转录相关复合物都说完了,是不是CHIP-seq的IP就到处为止了呢?要做CHIP-seq,我们的IP必然跟DNA有联系,这个联系可以是直接的,必然histone包围DNA,必然转录因子结合promoter,必然poly II 复合物结合DNA,那么其它核内蛋白,难道就不跟DNA相接触了吗?那么这是第8个协作任务, 人的2万多个基因里面,还有哪些是核内蛋白,一个完整的列表。
13、说到了核内蛋白,那么它是不是就一定会跟DNA接触了?如果接触,IP抓取这个蛋白的同时就能抓取DNA序列,就有CHIP-seq数据来进行下游分析。如果这个蛋白跟DNA接触了,它是不是就落入了转录因子的定义呢?转录因子分成general和specific的,转录因子有3个区域,看起前面的示意图。什么样的蛋白才有跟DNA接触的生物学基础呢?参照转录因子的3个结构共性。这是第9个协作任务。
14、那我们再谈一谈基因研究的问题,基因必然有promoter区域,有可能被某些specific的TF结合,这个就很值得研究,有数据库。gene的3'UTR区可以预测它是否被miRNA调控。基因必然可以被knock -down or OFF,或者overexpression,这个叫做基因的permutation,那么,哪些基因被研究过,被做过permutation呢?这是第10个协作任务。
15、说到基因的permutation,那么它在不同的tissue,不同的cell line,不同的disease状态,不同的处理,会有不同的后果。这就是大部分文章的终极意义。其中我上面提到的各种酶,各种转录因子,是优先被研究优先被做permutation的。
16、factorbook里面有encode计划的高标准TF,对人来说,有167个(http://www.factorbook.org/human/chipseq/tf/),那么它们的CHIP-seq数据的bam文件和peaks文件都可以拿到,TSS,genebody的profile图和热图,都可以拿到。这个是chipseq数据结果的理解,第11个协作。找到CHIP-seq的可视化图片,越多的类型越好。我会在两个月后给大家一个视频讲解。争取让大家都理解为什么要做CHIP-seq,已经做完CHIP--seq的结果有什么生物学意义
17、factorbook里面有encode计划的高标准TF,对人来说,有167个, 但是却有,837个实验数据,因为TF可以在不同的细胞系发挥作用,大家可以检查两个细胞系看看差异到底有多大,有一个大致的印象。这是第12个协作任务,会下载数据的,可以试一下。然后也是因为同一个TF,有多个不同商家的IP,这个问题就来了,IP的重要性不需要我多说了,这是第13个协作任务,搜集类似的关于IP的重要性的资料。
18、encode计划采用4种高通量测序技术来刻画了超过100种不同的细胞系或者组织内的全基因组范围内的基因调控元件信息。但是我们目前只关心CHIP-seq的技术手段的结果。所有数据从raw data形式的原始测序数据到比对后的信号文件以及分析好的有意的peaks文件都可以下载。下载方法见:http://www.bio-info-trainee.com/1825.html包括ENCODE官网下载,UCSC下载,ENSEMBL下载,broad研究所数据,IHEC存放的数据,还有GEO下载这6种形式!!!这是第14个协作任务,下载H3K4me3和H3K27me3的任意一种细胞系的peaks,然后看看它们的peaks的overlap情况。
19、第15个协作任务,上面我们谈到了encode计划涉及到的100种细胞系,那么有一个细胞系收集整理表格,有人已经做好,你们需要查询到,然后整理一份,跟大家讨论,我们要记住一些常见的细胞系的名字。(这个我手上有!)
20、熟悉一下3个数据库, http://cistrome.org/db/#/ http://www.genecards.org/http://biocc.hrbmu.edu.cn/GPA/AdvFuncSearchAction 其中GPA来看某个基因被干扰是否影响某个基因的表达,正反查询均可。 cistrome查询某个chip-seq是否被做过,结果是怎么样的! genecard里面增加了调控原件这个信息,大家可以了解一下。 如果你了解了这3个数据,就做一个调研吧,这是第16个协作任务,EGR1和EZH2两个基因相互作用关系
原理




上面的箭头中所示综述 老大着重提了一下
要看的文章

review-depth and coverage是刘小乐的综述
- 1

- 2

- 3

流程图
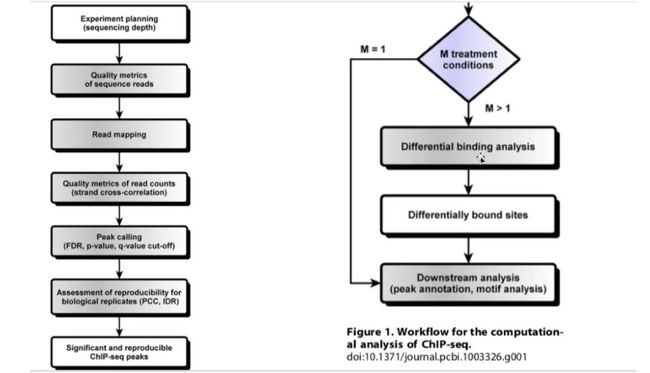


==划重点==
chip-seq的bam文件很大,因此要变成==bw==、bed、bedgraph格式,这些格式放在IGV里面方便看,bw文件看测序深度信息。比如知道某个位置比对上了10条reads,不需要知道这10条reads是A\T\C\G,因为他和参考基因组是一样的,因为不找变异,可能有一两个基因不一样,但是对chip-seq来说,没有任何意义,只需要这个位置如果比对上去的话,按测序深度就==+1==,所以我只需要知道测序深度就可以了。
-
老大说下面这个是讲的最详细的
 image-20191014225607361
image-20191014225607361 -
双峰模型
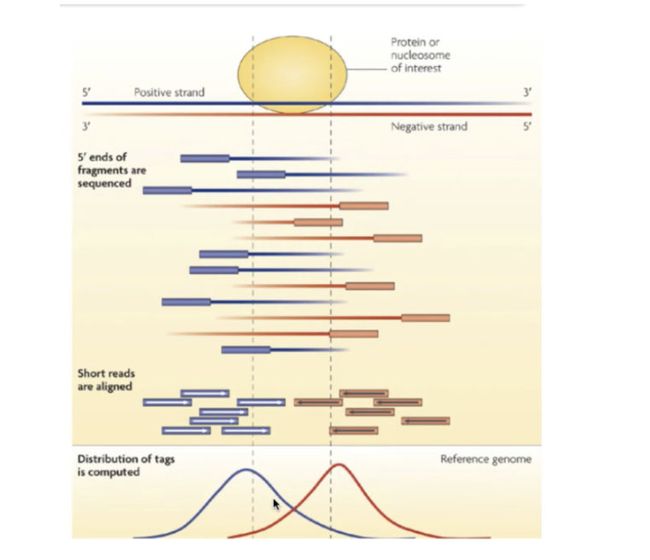 image-20191121211804979
image-20191121211804979 -
youtube上的视频可以看一下
 image-20191015081712176
image-20191015081712176
蛋白很难洗脱,因此可以做一个背景测序,如果背景都有peaks,那么就不是真正的peaked
- 实例


实战所需软件

实战数据
-
实战涉及文章==gse34520==
 image-20191015083023254
image-20191015083023254

表观调控之创建基于python2的conda环境
# wget https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
conda create -n epi python=2
conda info --envs
source activate epi
# 可以用search先进行检索
conda search trim_galore
## 保证所有的软件都是安装在 wes 这个环境下面
conda install -y sra-tools
conda install -y trim-galore samtools
conda install -y deeptools homer meme
conda install -y macs2 bowtie bowtie2
需要注意的是创建环境,我这里使用的代码是: conda create -n epi python=2
默认安装的是:
ca-certificates: 2019.8.28-0 defaults
certifi: 2019.9.11-py27_0 defaults
libcxx: 4.0.1-hcfea43d_1 defaults
libcxxabi: 4.0.1-hcfea43d_1 defaults
libedit: 3.1.20181209-hb402a30_0 defaults
libffi: 3.2.1-h475c297_4 defaults
ncurses: 6.1-h0a44026_1 defaults
pip: 19.2.3-py27_0 defaults
python: 2.7.16-h97142e2_7 defaults
readline: 7.0-h1de35cc_5 defaults
setuptools: 41.4.0-py27_0 defaults
sqlite: 3.30.0-ha441bb4_0 defaults
tk: 8.6.8-ha441bb4_0 defaults
wheel: 0.33.6-py27_0 defaults
zlib: 1.2.11-h1de35cc_3 defaults
公共数据获取

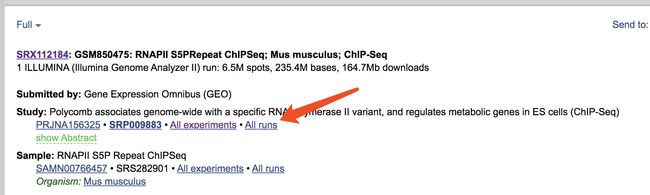
geo官网搜索gse34518


SRR391032
SRR391033
SRR391034
SRR391035
SRR391036
SRR391037
SRR391040
SRR391041
SRR391042
SRR391043
SRR391044
SRR391045
SRR391046
SRR391047
SRR391048
SRR391049
SRR391050
SRR391039
SRR391038
prefetch=/home/jianmingzeng/biosoft/sratoolkit/sratoolkit.2.8.2-1-centos_linux64/bin/prefetch
source activate chipseq
prefetch=prefetch
# cat srr.list |while read id;do (nohup $prefetch $id -X 100G & );done
mkdir -p ~/project/epi/
cd ~/project/epi/
mkdir {sra,raw,clean,align,peaks,motif,qc}
cd sra
## vim 或者cat命令创建 srr.list 文件。
cat srr.list |while read id;do ( nohup $prefetch $id & );done
## 默认下载目录:~/ncbi/public/sra/
ls -lh ~/ncbi/public/sra/
## 下载耗时,自行解决,学员使用现成数据:/public/project/epi/Chipseq-OS25_Esc/OS25_Esc/sra
## 假如提前下载好了数据。
cd ~/project/epi/
ln -s /public/project/epi/Chipseq-OS25_Esc/OS25_Esc/sra sra

上面的RunInfo Table得到如下图
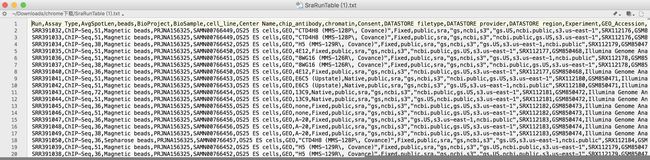
下面是视频中的
把第一列的分隔符转换为\n,换行后就知道每一列是什么了
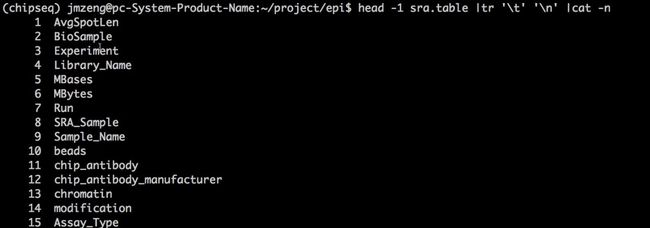
疑问,下面这个图片中的要制作的config文件中的第一列和我们上面下载的这个RunInfo Table什么关系?
后来明白了
制作config
cut -f 4,7 sra.table |cut -d":" -f 2 |sed 's/ChIPSeq//g' | sed 's/MockIP//g'|sed 's/^ //' |tr ' ' '_' |perl -alne '{$h{$F[0]}++ if exists $h{$F[0]}; $h{$F[0]}=1 unless exists $h{$F[0]};print "$F[0]$h{$F[0]}\t$F[1]"}' > config

## 下面需要用循环
cd ~/project/epi/
source activate chipseq
dump='/home/jianmingzeng/biosoft/sratoolkit/sratoolkit.2.8.2-1-centos_linux64/bin/fastq-dump'
dump=fastq-dump
analysis_dir=raw
## 下面用到的 config 文件,就是上面自行制作的。
cat config|while read id;
do echo $id
arr=($id)
srr=${arr[1]}
sample=${arr[0]}
# 单端测序数据的sra转fasq####
nohup $dump -A $sample -O $analysis_dir --gzip --split-3 sra/$srr.sra &
done
我觉得这个代码很重要。
- ==疑问,下面的这个$dump是上面两个dump中的哪一个呢?==
[图片上传失败...(image-54c9b-1574342586095)]
427M Jul 28 15:25 Control_1.fastq.gz
527M Jul 28 15:26 Control_2.fastq.gz
507M Jul 28 15:26 H2Aub1_1.fastq.gz
843M Jul 28 15:28 H2Aub1_2.fastq.gz
881M Jul 28 15:28 H3K36me3_1.fastq.gz
858M Jul 28 15:29 RNAPII_S2P_1.fastq.gz
326M Jul 28 15:25 RNAPII_S2P_2.fastq.gz
489M Jul 28 15:26 RNAPII_S2P_3.fastq.gz
283M Jul 28 15:25 RNAPII_S5PRepeat_1.fastq.gz
745M Jul 28 15:27 RNAPII_S5P_2.fastq.gz
533M Jul 28 15:26 RNAPII_S7P_1.fastq.gz
393M Jul 28 15:25 RNAPII_S7P_2.fastq.gz
266M Jul 28 15:25 Ring1B_1.fastq.gz
274M Jul 28 15:25 Ring1B_2.fastq.gz
目前遇到的问题
- ==红色画框这个地方还是不懂 得自己跑一下理解一下==,==蓝色、红色、黑色,到底哪个是命令呢?==
- 视频中提到了下面
-p 5的问题,样本19个,电脑就爆掉了。关于这个问题,也不太懂,后面看看能不能明白 - 这里面的参数问题,需要搜集一下

- 有知识背景在后面

- 单端测序/双端测序
- 可以去看外显子的流程
cd ~/project/epi/align
## 相对目录需要理解
bin_bowtie2='/home/jianmingzeng/biosoft/bowtie/bowtie2-2.2.9/bowtie2'
bin_bowtie2=bowtie2
bowtie2_index="/home/jianmingzeng/reference/index/bowtie/mm10"
bowtie2_index=/public/reference/index/bowtie/mm10
## 一定要搞清楚自己的bowtie2软件安装在哪里,以及自己的索引文件在什么地方!!!
ls ../clean/*gz |while read id; #id 代表前面../clean/*gz
do
file=$(basename $id )#basename就是去到了../clean/*gz中的*gz
sample=${file%%.*}#%%就是从右向左匹配,匹配到最左边的‘.’,然后这个‘.’及右边的
echo $file $sample
## 比对过程3分钟一个样本
$bin_bowtie2 -p 5 -x $bowtie2_index -U $id | samtools sort -O bam -@ 5 -o - > ${sample}.bam
done
- 上面的shell脚本得到的结果如下图
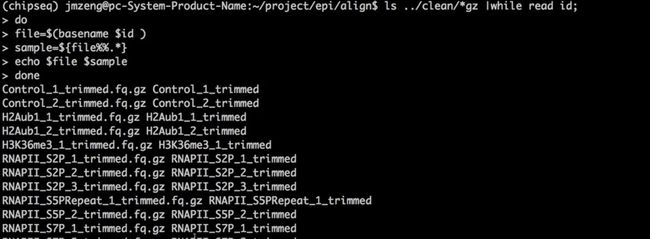
*gz就是Control_1_trimmed.fq.gz这一列
${file%%.*}就是去掉了‘.fq.gz’,留下了Control_1_trimmed
- 加nohup前

-
加nohup后


374M Jul 28 16:50 Control_1_trimmed.bam
469M Jul 28 16:50 Control_2_trimmed.bam
502M Jul 28 16:50 H2Aub1_1_trimmed.bam
767M Jul 28 16:50 H2Aub1_2_trimmed.bam
834M Jul 28 16:50 H3K36me3_1_trimmed.bam
731M Jul 28 16:28 RNAPII_S2P_1_trimmed.bam
302M Jul 28 16:29 RNAPII_S2P_2_trimmed.bam
483M Jul 28 16:32 RNAPII_S2P_3_trimmed.bam
218M Jul 28 16:33 RNAPII_S5PRepeat_1_trimmed.bam
609M Jul 28 16:36 RNAPII_S5P_2_trimmed.bam
416M Jul 28 16:38 RNAPII_S7P_1_trimmed.bam
309M Jul 28 16:39 RNAPII_S7P_2_trimmed.bam
238M Jul 28 16:40 Ring1B_1_trimmed.bam
239M Jul 28 16:41 Ring1B_2_trimmed.bam
- 对bam文件进行QC
cd ~/project/epi/align
ls *.bam |xargs -i samtools index {}
ls *.bam | while read id ;do (nohup samtools flagstat $id > $(basename $id ".bam").stat & );done
bai.bam是index
比对成功率都挺好的:
Control_1_trimmed.stat:7438540 + 0 mapped (88.03% : N/A)
Control_2_trimmed.stat:7221781 + 0 mapped (86.40% : N/A)
H2Aub1_1_trimmed.stat:8969578 + 0 mapped (97.40% : N/A)
H2Aub1_2_trimmed.stat:13229916 + 0 mapped (97.53% : N/A)
H3K36me3_1_trimmed.stat:11737310 + 0 mapped (98.89% : N/A)
Ring1B_1_trimmed.stat:4634240 + 0 mapped (93.59% : N/A)
Ring1B_2_trimmed.stat:4646919 + 0 mapped (93.85% : N/A)
RNAPII_S2P_1_trimmed.stat:25018794 + 0 mapped (97.26% : N/A)
RNAPII_S2P_2_trimmed.stat:6112834 + 0 mapped (95.00% : N/A)
RNAPII_S2P_3_trimmed.stat:8675514 + 0 mapped (96.99% : N/A)
RNAPII_S5P_2_trimmed.stat:12182274 + 0 mapped (98.17% : N/A)
RNAPII_S5PRepeat_1_trimmed.stat:4163763 + 0 mapped (82.81% : N/A)
RNAPII_S7P_1_trimmed.stat:6386269 + 0 mapped (80.90% : N/A)
RNAPII_S7P_2_trimmed.stat:5971178 + 0 mapped (82.66% : N/A)
合并bam文件 ==samtools merge==
因为一个样品分成了多个lane进行测序,所以在进行peaks calling的时候,需要把bam进行合并。
## 如果不用循环:
## samtools merge control.merge.bam Control_1_trimmed.bam Control_2_trimmed.bam
## 通常我们用批处理。
cd ~/project/epi/
mkdir mergeBam
source activate chipseq
cd ~/project/epi/align
ls *.bam|sed 's/_[0-9]_trimmed.bam//g' |sort -u |while read id;do samtools merge ../mergeBam/$id.merge.bam $id*.bam ;done
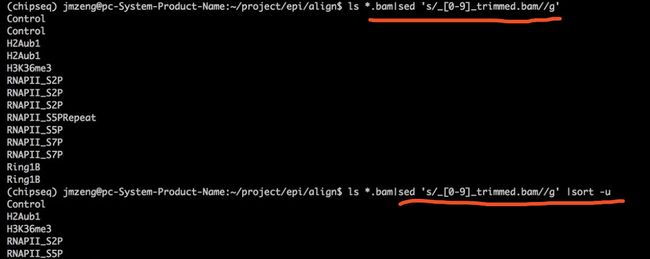
- 看一下这里面
ls *.bam|sed 's/_[0-9]_trimmed.bam//g' |sort -u |while read id;do samtools merge ../mergeBam/$id.merge.bam $id*.bam ;done
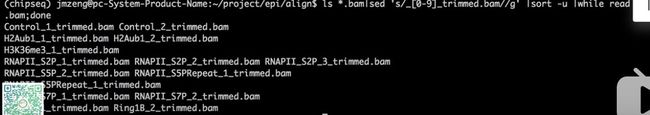
得到全新的bam文件如下:
847M Jul 28 17:05 Control.merge.bam
1.3G Jul 28 17:06 H2Aub1.merge.bam
834M Jul 28 17:06 H3K36me3.merge.bam
1.5G Jul 28 17:08 RNAPII_S2P.merge.bam
831M Jul 28 17:09 RNAPII_S5P.merge.bam
218M Jul 28 17:09 RNAPII_S5PRepeat.merge.bam
722M Jul 28 17:09 RNAPII_S7P.merge.bam
472M Jul 28 17:10 Ring1B.merge.bam
14个fq测序数据只剩下8个样本啦。(我下载的时候漏掉了2个sra文件,也就是漏掉了一个样本。)
- 有pcr重复的时候会发现一条reads出现两三次
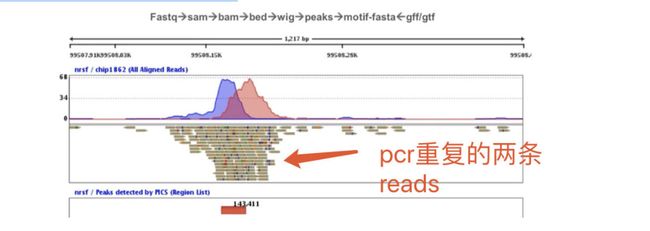
去重复的软件samtools和picard
菜鸟团搜索重复,有两篇,如下图
去重复就是去起始终止点都一样的

假如需要去除PCR重复
- ==这里面的stat和flagstat是什么来着==
cd ~/project/epi/mergeBam
source activate chipseq
ls *merge.bam | while read id ;do (nohup samtools markdup -r $id $(basename $id ".bam").rmdup.bam & );done
ls *.rmdup.bam |xargs -i samtools index {}
ls *.rmdup.bam | while read id ;do (nohup samtools flagstat $id > $(basename $id ".bam").stat & );done
去除PCR重复前后比较:可以看到去除重复以后文件大小还是改变了
847M Jul 28 17:05 Control.merge.bam
753M Jul 28 17:11 Control.merge.rmdup.bam
1.3G Jul 28 17:06 H2Aub1.merge.bam
1.1G Jul 28 17:12 H2Aub1.merge.rmdup.bam
834M Jul 28 17:06 H3K36me3.merge.bam
793M Jul 28 17:11 H3K36me3.merge.rmdup.bam
1.5G Jul 28 17:08 RNAPII_S2P.merge.bam
1.2G Jul 28 17:12 RNAPII_S2P.merge.rmdup.bam
831M Jul 28 17:09 RNAPII_S5P.merge.bam
568M Jul 28 17:11 RNAPII_S5P.merge.rmdup.bam
218M Jul 28 17:09 RNAPII_S5PRepeat.merge.bam
212M Jul 28 17:11 RNAPII_S5PRepeat.merge.rmdup.bam
722M Jul 28 17:09 RNAPII_S7P.merge.bam
618M Jul 28 17:11 RNAPII_S7P.merge.rmdup.bam
472M Jul 28 17:10 Ring1B.merge.bam
427M Jul 28 17:11 Ring1B.merge.rmdup.bam
使用macs2进行找peaks
macs2包含一系列的子命令,其中最主要的就是callpeak, 官方提供了使用实例
macs2 callpeak -t ChIP.bam -c Control.bam -f BAM -g hs -n test -B -q 0.01
一般而言,我们照葫芦画瓢,按照这个实例替换对应部分就行了,介绍一下各个参数的意义
- -t: 实验组的输出结果
- -c: 对照组的输出结果
- -f: -t和-c提供文件的格式,可以是”ELAND”, “BED”, “ELANDMULTI”, “ELANDEXPORT”, “ELANDMULTIPET” (for pair-end tags), “SAM”, “BAM”, “BOWTIE”, “BAMPE” “BEDPE” 任意一个。如果不提供这项,就是自动检测选择。
- -g: 基因组大小, 默认提供了hs, mm, ce, dm选项, 不在其中的话,比如说拟南芥,就需要自己提供了。
- -n: 输出文件的前缀名
- -B: 会保存更多的信息在bedGraph文件中,如fragment pileup, control lambda, -log10pvalue and -log10qvalue scores
- -q: q值,也就是最小的PDR阈值, 默认是0.05。q值是根据p值利用BH计算,也就是多重试验矫正后的结果。
- -p: 这个是p值,指定p值后MACS2就不会用q值了。
- -m: 和MFOLD有关,而MFOLD和MACS预构建模型有关,默认是5:50,MACS会先寻找100多个peak区构建模型,一般不用改,因为你很大概率上不会懂。
所以我这里给学徒讲解的实战代码是:
cd ~/project/epi/mergeBam
source activate chipseq
ls *merge.bam |cut -d"." -f 1 |while read id;
do
if [ ! -s ${id}_summits.bed ];
then
echo $id
nohup macs2 callpeak -c Control.merge.bam -t $id.merge.bam -f BAM -B -g mm -n $id --outdir ../peaks 2> $id.log &
fi
done
mkdir dup
mv *rmdup* dup/
cd dup/
ls *.merge.rmdup.bam |cut -d"." -f 1 |while read id;
do
if [ ! -s ${id}_rmdup_summits.bed ];
then
echo $id
nohup macs2 callpeak -c Control.merge.rmdup.bam -t $id.merge.rmdup.bam -f BAM -B -g mm -n ${id}_rmdup --outdir ../peaks 2> $id.log &
fi
done
其实上面的-B 参数意义也不大,得到的bedgraph文件没啥用。
老大在菜鸟团里有wig、bigwig、bedgraph文件详解
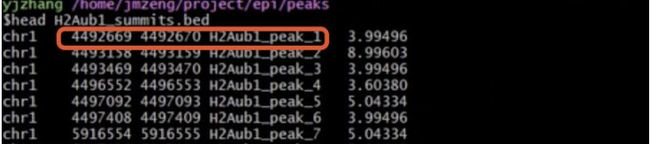
得到的bed格式的peaks文件的行数如下:
0 Control_summits.bed
1102 H2Aub1_summits.bed
89739 H3K36me3_summits.bed
27705 Ring1B_summits.bed
20043 RNAPII_S2P_summits.bed
38643 RNAPII_S5PRepeat_summits.bed
61805 RNAPII_S5P_summits.bed
72498 RNAPII_S7P_summits.bed
0 Control_rmdup_summits.bed
1102 H2Aub1_rmdup_summits.bed
89739 H3K36me3_rmdup_summits.bed
27705 Ring1B_rmdup_summits.bed
20043 RNAPII_S2P_rmdup_summits.bed
38643 RNAPII_S5PRepeat_rmdup_summits.bed
61805 RNAPII_S5P_rmdup_summits.bed
72326 RNAPII_S7P_rmdup_summits.bed
因为MockIP是control,所以它自己跟自己比较,肯定是没有peaks的。
值得注意的是S5P并不是一个样本多个lane,而是本身样本有重复,其实是需要分开的。
而且可以看到是否去除PCR重复,对找到的peaks数量没有影响。
而且很有趣的是我前几个月处理这个数据集的时候使用的过滤低质量reads参数是短于 35bp的全部丢弃,现在是短于25bp的全部抛弃,导致了得到的peaks从数量上千差别不小。
- ==看视频中的老大提出的问题,比如control组,从merge.bam到merge.rmdup.bam少了近100bp,但是两组中的peaks却没有减少。==
使用deeptool是进行可视化
下面的文字摘抄自生信技能树论坛:https://vip.biotrainee.com/d/226 不过代码纯粹是我自己手打。
deeptools提供bamCoverage和bamCompare进行格式转换,为了能够比较不同的样本,需要对先将基因组分成等宽分箱(bin),统计每个分箱的read数,最后得到描述性统计值。对于两个样本,描述性统计值可以是两个样本的比率,或是比率的log2值,或者是差值。如果是单个样本,可以用SES方法进行标准化。
bamCoverage的基本用法
source activate chipseq
bamCoverage -e 170 -bs 10 -b ap2_chip_rep1_2_sorted.bam -o ap2_chip_rep1_2.bw
# ap2_chip_rep1_2_sorted.bam是前期比对得到的BAM文件
得到的bw文件就可以送去IGV/Jbrowse进行可视化。 这里的参数仅使用了-e/--extendReads和-bs/--binSize即拓展了原来的read长度,且设置分箱的大小。其他参数还有
-
--filterRNAstrand {forward, reverse}: 仅统计指定正链或负链 -
--region/-r CHR:START:END: 选取某个区域统计 -
--smoothLength: 通过使用分箱附近的read对分箱进行平滑化
如果为了其他结果进行比较,还需要进行标准化,deeptools提供了如下参数:
-
--scaleFactor: 缩放系数 -
--normalizeUsingRPKMReads: Per Kilobase per Million mapped reads (RPKM)标准化 -
--normalizeTo1x: 按照1x测序深度(reads per genome coverage, RPGC)进行标准化 -
--ignoreForNormalization: 指定那些染色体不需要经过标准化
如果需要以100为分箱,并且标准化到1x,且仅统计某一条染色体区域的正链,输出格式为bedgraph,那么命令行可以这样写
bamCoverage -e 170 -bs 100 -of bedgraph -r Chr4:12985884:12997458 --normalizeTo1x 100000000 -b 02-read-alignment/ap2_chip_rep1_1_sorted.bam -o chip.bedgraph
bamCompare和bamCoverage类似,只不过需要提供两个样本,并且采用SES方法进行标准化,于是多了--ratio参数。
首先把bam文件转为bw文件,详情:http://www.bio-info-trainee.com/1815.html
cd ~/project/epi/mergeBam
source activate chipseq
ls *.bam |xargs -i samtools index {}
ls *.bam |while read id;do
nohup bamCoverage --normalizeUsing CPM -b $id -o ${id%%.*}.bw &
done
cd dup
ls *.bam |xargs -i samtools index {}
ls *.bam |while read id;do
nohup bamCoverage --normalizeUsing CPM -b $id -o ${id%%.*}.rm.bw & #加了一个rm,变为rm.bw
done
==上面是两套分析方案,因为看文章不知道rmdup后有没有区别==
==上面由于测的样本比较多,可能会误以为某个位置是peaks,所以需要normalizeUsing,rpkm需要基因的长度,而这里面并没有用基因的长度,所以用cpm==
==上面注意一定要有index==
==还有一个问题,是否去除多比对reads==,==有一些reads会比对到多个地方,不同的比对工具有不同的比对策略==
xargs -i samtools index {} #一直不太懂
做成转入IGV需要的样子
awk '{print $1":"$2"-"$3}' + 文件名.bed

- ==但是如果bed文件仍然太大的话,就可以用bw文件载入IGV,有同样的效果==



- 去掉假阳性才能得到真正的peaks
- 目的是能找到peaks,知道位置
- TSS
查看TSS附件信号强度:可以用==deeptools==画
## 首先对单一样本绘图:
## both -R and -S can accept multiple files
mkdir -p ~/project/epi/tss
cd ~/project/epi/tss
computeMatrix reference-point --referencePoint TSS -p 15 \
-b 10000 -a 10000 \
-R /public/annotation/CHIPseq/mm10/ucsc.refseq.bed \
-S /home/jmzeng/project/epi/mergeBam/H2Aub1.bw \
--skipZeros -o matrix1_test_TSS.gz \
--outFileSortedRegions regions1_test_genes.bed
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_test_TSS.gz -out test_Heatmap.png
plotHeatmap -m matrix1_test_TSS.gz -out test_Heatmap.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_test_TSS.gz -out test_Profile.png
plotProfile -m matrix1_test_TSS.gz -out test_Profile.pdf --plotFileFormat pdf --perGroup --dpi 720
### 如果要批处理 ,需要学习好linux命令。
首先画10K附近
bed=/public/annotation/CHIPseq/mm10/ucsc.refseq.bed
for id in /home/jmzeng/project/epi/mergeBam/*bw ;
do
echo $id
file=$(basename $id )
sample=${file%%.*}
echo $sample
computeMatrix reference-point --referencePoint TSS -p 15 \
-b 10000 -a 10000 \
-R $bed \
-S $id \
--skipZeros -o matrix1_${sample}_TSS_10K.gz \
--outFileSortedRegions regions1_${sample}_TSS_10K.bed
# 输出的gz为文件用于plotHeatmap, plotProfile
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_${sample}_TSS_10K.gz -out ${sample}_Heatmap_10K.png
plotHeatmap -m matrix1_${sample}_TSS_10K.gz -out ${sample}_Heatmap_10K.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_${sample}_TSS_10K.gz -out ${sample}_Profile_10K.png
plotProfile -m matrix1_${sample}_TSS_10K.gz -out ${sample}_Profile_10K.pdf --plotFileFormat pdf --perGroup --dpi 720
done
使用命令批量提交:nohup bash 10k.sh 1>10k.log &
然后画2K的
bed=/public/annotation/CHIPseq/mm10/ucsc.refseq.bed
for id in /home/jmzeng/project/epi/mergeBam/*bw ;
do
echo $id
file=$(basename $id )
sample=${file%%.*}
echo $sample
computeMatrix reference-point --referencePoint TSS -p 15 \
-b 2000 -a 2000 \
-R $bed \
-S $id \
--skipZeros -o matrix1_${sample}_TSS_2K.gz \
--outFileSortedRegions regions1_${sample}_TSS_2K.bed
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Heatmap_2K.png
plotHeatmap -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Heatmap_2K.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Profile_2K.png
plotProfile -m matrix1_${sample}_TSS_2K.gz -out ${sample}_Profile_2K.pdf --plotFileFormat pdf --perGroup --dpi 720
done
使用命令批量提交:nohup bash 2k.sh 1>2k.log &
还可以给多个bed文件来绘图,还可以画genebody的图,因为原理一样,我就不做过多介绍啦。
于 2018年10月06日23:07:29 更新
我还是准备把这个代码发出来:
```
#!/bin/bash
source activate chip
bed=/public/annotation/CHIPseq/mm10/ucsc.refseq.bed
for id in /home/llwu/jmzeng/align/*.rmdup.bw ;
do
echo $id
file=$(basename $id )
sample=${file%%.*}
echo $sample
computeMatrix scale-regions -p 5 \
-b 3000 -a 3000 \
-R $bed \
-S $id \
--regionBodyLength 15000 --skipZeros \
-o matrix1_${sample}_genebody.gz \
--outFileSortedRegions regions1_${sample}_genebody.bed
## both plotHeatmap and plotProfile will use the output from computeMatrix
plotHeatmap -m matrix1_${sample}_genebody.gz -out ${sample}_Heatmap_genebody.png
plotHeatmap -m matrix1_${sample}_genebody.gz -out ${sample}_Heatmap_genebody.pdf --plotFileFormat pdf --dpi 720
plotProfile -m matrix1_${sample}_genebody.gz -out ${sample}_Profile_genebody.png
plotProfile -m matrix1_${sample}_genebody.gz -out ${sample}_Profile_genebody.pdf --plotFileFormat pdf --perGroup --dpi 720
done
```
同样是使用命令批量提交:`nohup bash genebody.sh 1>genebody.log &`
上面的批量代码其实就是为了统计全基因组范围的peak在基因特征的分布情况,也就是需要用到computeMatrix计算,用plotHeatmap以热图的方式对覆盖进行可视化,用plotProfile以折线图的方式展示覆盖情况。
computeMatrix具有两个模式:scale-region和reference-point。前者用来信号在一个区域内分布,后者查看信号相对于某一个点的分布情况。无论是那个模式,都有有两个参数是必须的,-S是提供bigwig文件,-R是提供基因的注释信息。还有更多个性化的可视化选项。
使用R包对找到的peaks文件进行注释
bedPeaksFile = '8WG16_summits.bed';
bedPeaksFile
## loading packages
require(ChIPseeker)
require(TxDb.Mmusculus.UCSC.mm10.knownGene)
txdb <- TxDb.Mmusculus.UCSC.mm10.knownGene
require(clusterProfiler)
peak <- readPeakFile( bedPeaksFile )
keepChr= !grepl('_',seqlevels(peak))
seqlevels(peak, pruning.mode="coarse") <- seqlevels(peak)[keepChr]
peakAnno <- annotatePeak(peak, tssRegion=c(-3000, 3000),
TxDb=txdb, annoDb="org.Mm.eg.db")
peakAnno_df <- as.data.frame(peakAnno)
可以载入IGV看看效果,检测软件找到的peaks是否真的合理,还可以配合rmarkdown来出自动化报告。
也可以使用其它代码进行下游分析; https://github.com/jmzeng1314/NGS-pipeline/tree/master/CHIPseq
peaks相关基因集的注释
- ==根据peaks的坐标和基因的坐标进行注释==,把peaks坐标和基因的坐标通过overlap来看这个peaks离哪个基因最近,以及在什么地方
都是得到感兴趣基因集,然后注释,分析方法等同于GEO数据挖掘课程或者转录组下游分析: https://github.com/jmzeng1314/GEO (有配套视频,就不多说了这里)
homer软件来寻找motif
这个软件安装当初特别麻烦: https://github.com/jmzeng1314/NGS-pipeline/blob/master/CHIPseq/step8-Homer-findMotif.sh
但是现在有了conda,一句话搞定:conda install -c bioconda homer , 找到自己安装的homer,然后使用其附带的配置脚本来下载数据库咯。
perl ~/miniconda3/envs/chipseq/share/homer-4.9.1-5/configureHomer.pl -install mm10
ls -lh ~/miniconda3/envs/chipseq/share/homer-4.9.1-5/data/
## 我们上游分析是基于mm10找到的peaks文件
## 数据库下载取决于网速咯
## 下载成功后会多出 ~/miniconda3/envs/chipseq/share/homer-4.9.1-5/data/genomes/mm9/ 文件夹, 共 4.9G
## 这个文件夹取决于你把homer这个软件安装到了什么地方。
## 或者用下面代码安装:
cd ~/biosoft
mkdir homer && cd homer
wget http://homer.salk.edu/homer/configureHomer.pl
perl configureHomer.pl -install
perl configureHomer.pl -install hg19
homer软件找motif整合了两个方法,包括依赖于数据库的查询,和de novo的推断,都是读取ChIP-seq数据上游分析得到的bed格式的peaks文件。
运行homer软件
但是使用起来很简单:http://homer.ucsd.edu/homer/ngs/peakMotifs.html
cd ~/project/epi/motif
for id in /home/jmzeng/project/epi/peaks/*.bed;
do
echo $id
file=$(basename $id )
sample=${file%%.*}
echo $sample
awk '{print $4"\t"$1"\t"$2"\t"$3"\t+"}' $id >homer_peaks.tmp
findMotifsGenome.pl homer_peaks.tmp mm10 ${sample}_motifDir -len 8,10,12
annotatePeaks.pl homer_peaks.tmp mm10 1>${sample}.peakAnn.xls 2>${sample}.annLog.txt
done
#上面是用mm10注释
#上面是$sample,就仅仅是文件名
上面的代码是可以先echo出来,就是进入cd ~/project/epi/motif 以后
for id in /home/jmzeng/project/epi/peaks/*.bed;
do
echo $id
file=$(basename $id )
sample=${file%%.*}
echo $sample


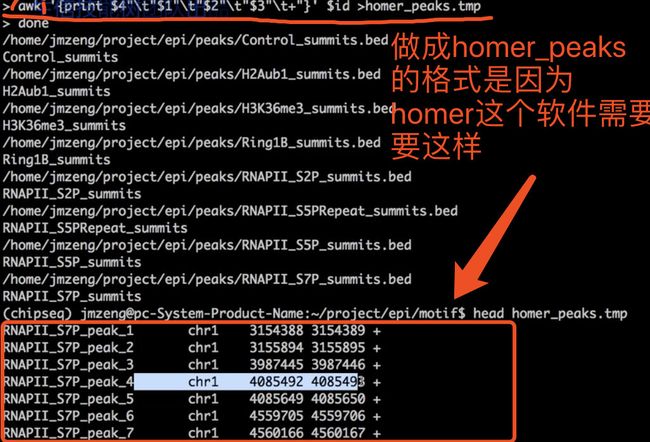
把上面的代码保存为脚本runMotif.sh,然后运行:nohup bash runMotif.sh 1>motif.log &
不仅仅找了motif,还顺便把peaks注释了一下。得到的后缀为peakAnn.xls 的文件就可以看到和使用R包注释的结果是差不多的。
还可以使用meme来找motif,需要通过bed格式的peaks的坐标来获取fasta序列。MEME,链接:http://meme-suite.org/
- 有bed文件就能得到fasta

- ==通过下面这个包(有bed文件)这种方式得到fa文件,就是通过坐标得到序列==

其它高级分析
比如可以 比较不同的peaks文件,代码见:https://github.com/jmzeng1314/NGS-pipeline/blob/master/CHIPseq/step6-ChIPpeakAnno-Venn.R
- 当然了,本教程讲解的是单端测序数据的处理,如果是双端测序,里面的很多参数是需要修改的。
- 不过,只要你完整的看完了我前面的流程,掌握了linux和R,以及必备的基础生物信息学知识,我相信你肯定能hold住双端测序数据的学习啦。
- 本来以为我把ChIP-seq教程写完了: 一不小心就把ChIP-seq数据分析教程给写完了
- 结果忘记了还有视频这回事,赶紧录制了,同样的B站免费送给大家看,但是不提供售后,网易云课堂也有视频: 把我的视频听2遍以上再发问: https://study.163.com/course/introduction/1005833006.htm
shell流程
直接上代码,我就不解释了,感兴趣的直接看视频教程
fq1=$1
fq2=$2
sample=$3
analysis_dir=$4
###############################################
source activate chip
bowtie2_index=/public/reference/index/bowtie/mm10
mkdir -p {sra,raw,clean,align,peaks,motif,qc}
###############################################
## step0: keep fq.gz for all raw fastq files.
if [[ $fq1 =~ ".fastq.gz" ]];then
r_fq1=$analysis_dir/raw/$(basename ${fq1/.fastq.gz/.fq.gz} )
r_fq2=$analysis_dir/raw/$(basename ${fq2/.fastq.gz/.fq.gz} )
ln -s $fq1 $r_fq1
ln -s $fq2 $r_fq2
echo 'match'
else
echo "not match"
r_fq1=$analysis_dir/raw/$(basename $fq1)
r_fq2=$analysis_dir/raw/$(basename $fq2)
ln -s $fq1 $r_fq1
ln -s $fq2 $r_fq2
fi
fq1=$(basename $r_fq1)
fq2=$(basename $r_fq2)
## step1: run fastqc for raw fastq files
if [ ! -f $analysis_dir/raw/${fq1/.fq.gz/_fastqc.html} ];then
fastqc $r_fq1
else
echo "skill fastqc for $fq1"
fi
if [ ! -f $analysis_dir/raw/${fq2/.fq.gz/_fastqc.html} ];then
fastqc $r_fq2
else
echo "skill fastqc for $fq2"
fi
## step2: run trim_galore
START_TIME=`date +%s`
if [ ! -f $analysis_dir/clean/${fq2}_trimming_report.txt ];then
trim_galore -q 25 --phred33 --length 35 -e 0.1 --stringency 4 --paired -o $analysis_dir/clean $r_fq1 $r_fq2
else
echo "skill trim_galore for $fq2"
fi
END_TIME=`date +%s`
EXECUTING_TIME=`expr $END_TIME - $START_TIME`
echo $EXECUTING_TIME
## step3: run fastqc for clean fastq files
if [ ! -f $analysis_dir/clean/${fq1/.fq.gz/_val_1_fastqc.html} ];then
fastqc $analysis_dir/clean/${fq1/.fq.gz/_val_1.fq.gz}
else
echo "skill fastqc for clean $analysis_dir/clean/${fq1/.fq.gz/_val_1.fq.gz} "
fi
if [ ! -f $analysis_dir/clean/${fq2/.fq.gz/_val_2_fastqc.html} ];then
fastqc $analysis_dir/clean/${fq2/.fq.gz/_val_2.fq.gz}
else
echo "skill fastqc for clean $analysis_dir/clean/${fq2/.fq.gz/_val_2.fq.gz} "
fi
## step4: map the clean reads in fq to reference genome by bowtie2
c_fq1=$analysis_dir/clean/${fq1/.fq.gz/_val_1.fq.gz}
c_fq2=$analysis_dir/clean/${fq2/.fq.gz/_val_2.fq.gz}
if [ ! -f $analysis_dir/align/${sample}.raw.bam ];then
bowtie2 -x $bowtie2_index -1 $c_fq1 -2 $c_fq2 |samtools sort -O bam -o - > $analysis_dir/align/${sample}.raw.bam
else
echo "skill alignment for $sample"
fi
samtools index $analysis_dir/align/${sample}.raw.bam
samtools flagstat $analysis_dir/align/${sample}.raw.bam > $analysis_dir/align/${sample}.raw.stat
sambamba markdup --overflow-list-size 600000 --tmpdir='./' \
-r $analysis_dir/align/${sample}.raw.bam $analysis_dir/align/${sample}.rmdup.bam
samtools index $analysis_dir/align/${sample}.rmdup.bam
## step5: call peaks by MACS2
macs2 callpeak -t $analysis_dir/align/${sample}.rmdup.bam -f BAM -g mm -n ${sample}_rmdup -q 0.01 --outdir $analysis_dir/peaks
macs2 callpeak -t $analysis_dir/align/${sample}.raw.bam -f BAM -g mm -n ${sample}_raw -q 0.01 --outdir $analysis_dir/peaks
## step6: bam2bed and bam2bw
bamCoverage -bs 10 -b $analysis_dir/align/${sample}.raw.bam -o $analysis_dir/align/${sample}.raw.bw
bamCoverage -bs 10 -b $analysis_dir/align/${sample}.rmdup.bam -o $analysis_dir/align/${sample}.rmdup.bw
bedtools bamtobed -i $analysis_dir/align/${sample}.raw.bam > $analysis_dir/align/${sample}.raw.bed
bedtools bamtobed -i $analysis_dir/align/${sample}.rmdup.bam > $analysis_dir/align/${sample}.rmdup.bed
- ==homer下载的文件里就有参考基因组,就有序列,有了这个序列就能做motif分析。motif分析需要在看教程,就是同样一个地方如果多次出现a,就把这个画大一点==

- 得到peaks的序列,有坐标就有序列
==得到的结果如下,就是某一个chip_seq的antibody倾向于结合这样的序列,如下图T、C、A这样的出现的比率比较高,有这样的序列,那么这个antibody所对应的transcript factor 或histone motification就会结合这样的区域==
-
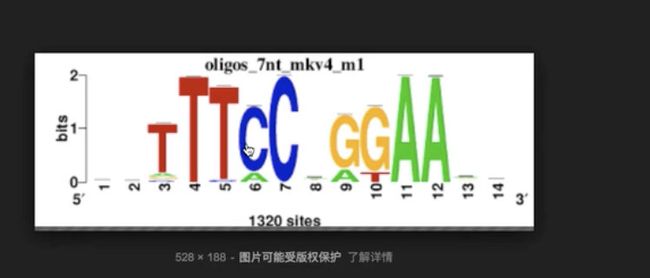 image-20191016183249373
image-20191016183249373 多个peaks文件
-
 image-20191016183820414
image-20191016183820414