系统功能注释和生物网络可视化
Spatial Analysis of Functional Enrichment(SAFE),功能富集空间分析
生物网络代表genes间的关系,但我们对这种网络功能组织的理解非常有限。这里,我描述spatial analysis of functional enrichment(SAFE),功能富集空间分析。SAFE在2D空间可视化网络并且度量跨越本地邻接功能富集的连续性分布,会产生相关联的功能列表和他们的相关性定位的map。我应用SAFE来注释啤酒酵母基因相互作用相似性网络和PPI网络,用GO terms。SAFE注释的基因网络匹配手工来源的注释,花的时间少,网络对噪音文件和生物信号敏感。使用SAFE对基因相互作用和化学基因组数据的整合揭示了囊泡介导的转运和对抗癌药物的抵抗之间的联系。这些结果显示SAFE在检查生物网络和理解他们的功能组织上的重要性。
--
INTRODUCTION
了解活细胞的功能组织对预测细胞在正常和疾病状态下的行为非常重要,这可以来设计有效的治疗策略来控制它们。出芽啤酒酵母对阐明细胞细胞的组织尤其有用,归因于扩展的分子相互作用网络的可获得性,可以匹配到物理,生物化学和表型间联系在几乎genome中的所有基因。然而,这些网络的功能注释,也就是说,决定那个生物功能可以代表每个网络,网络中的部分和哪些功能联系,他们如何和另外一个联系,是一个挑战,尤其是因为严格的统计学方法和可重复的工作流程的稀少。
生物网络的系统注释可以用三步阐明
首先,获得网络的综合map,来显示所有的nodes和他们之间的彼此联系,这个map可以由嵌于网络中的网络显示算法生成(2Dor3D),所有nodes的位置基于他们的连接性定位。
第二,收集可以用来定位所有nodes之间特征的独立的数据集,这种关系基于多样化参数(比如,细胞定位,对扰乱刺激的转录反应,突变表型等)。这种功能资源对酵母来说很容易获得,因为技术进步和很多人的贡献。
最后,应用自动的统计学程序叠加共功能数据到网络中并且鉴定功能一致的区域。已经有功能区域被发现,但是到现在为止,没有已有的方法来鉴定他们,相对于彼此定位并产生网络的功能地图,可以被生物学界理解,定量并且直观。
这些算法的主要作用是评估实验数据集,并确定网络连接支持的最有希望的候选基因。由于网络本身不是分析的重点,而只是独立的支持性证据,这些方法不能直接应用于综合性的注释任务。类似地,网络聚类算法可能被用来识别与已知的以及新颖的功能模块相对应的密集连接的节点组(Newman,2006)。然而,聚类忽视松散连接的节点,导致许多稀疏但功能一致的网络区域不被注意。另外,聚类将网络划分为离散的,并且在一些情况下是重叠的子网络,这些子网络必须被分别注释和整合,重新融合在一起,以提供网络的全局功能视图。由于功能注释的快速且可重现的整合尚未系统地实现,因此使用用于注释生物网络的聚类算法是不切实际的。
SAFE总概
SAFE通过计算和展示一系列功能属性来注释生物网络。SAFE首先产生网络2Dmap,用的是force-directed network layout算法或者从第三方软件输入比如cytoscape。在网络map中,nodes的定位基于反应网络拓扑异构性的forces 的平衡性进行定位,比如链接的nodes吸引彼此,而没链接的nodes排斥。
对于网络map中的每一个node来说,SAFE定义一个本地邻居,比如一系列nodes定位在一个特定的范围之内,但是不必直接链接。下图B。对于每一个邻居,SAFE计算一系列定量得分,每一个都相应于邻居的数值综合。
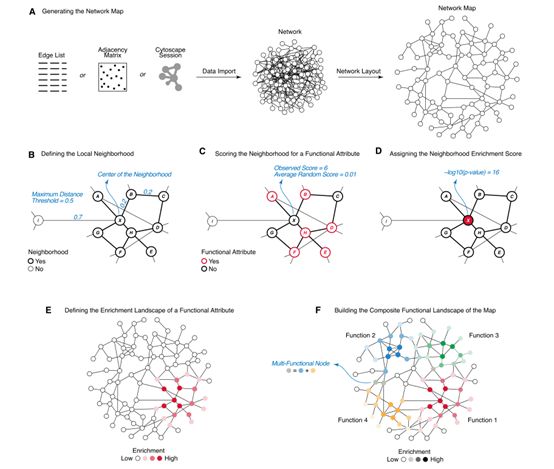
Benchmarking SAFE on the 酵母遗传相互作用相似性网络
为了检验SAFE方法,我应用它来注释酵母遗传相互作用相似性网络(GIS,genetic interaction similarity),使用的GO生物过程terms作为属性,并把这个结果与之前手工注释的进行比较。一个遗传相互作用是两个基因之间的表型联系,遗传相互作用是当双突变体的表型偏离两个单突变体的表型的预期组合时表型关系的两个基因。享有相似的基因相互作用的genes通常享有以共同的生物学功能并形成一个功能网络,并在在酵母基因组中更能有可能链接大多数的genes。一个高度保守的网络版本,包含2838个nodes,10016条边,通过一个彻底的,手工的程序基于原始研究进行注释,因此它对SAFE提供了一个很好的test。(这就是benchmarking)
在cytoscape中,使用spring-embedded network layout 生成GIS(genetic interaction similarit)map。SAFE度量4373个GO生物过程条目的本地富集,每一个都至少和一个酵母gene相关,揭示在size,shape和GO term富集landscape的变化,fig2B,C,D。GO term的大多数84%只在是个或更少个genes邻居内富集,显示了这些term在网络中太小或太稀疏分布以至于没有功能组织的信息。Fig2b。保留的GO TERMS富集在多于十个的邻居,但是在他们的富集中变化有空间分布变化:12%的GO terms(506 of 4373),有区域特异性就像它们在网络的单个区域有富集单个peak。Fig2c。,4%的GO terms在不同的网络区域有两个或多个peaks多区域分布。多个peack的出现显示了每一个多区域条目包含了几个genes的亚类,分别定位在网络中,这样可以功能区分。值得注意的是,每个亚类显示被至少一个区域特异地图覆盖,这显示了区域特异GO terms足够注释整个网络,作为结果,稀少的和多区域的条目不会在下一步进行分析。
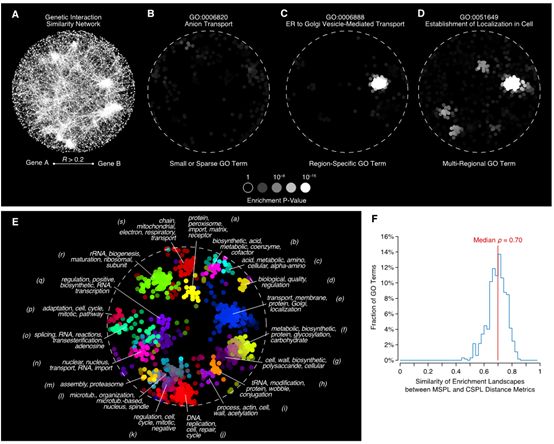

因为506个区域特异性GO条目中的很多都匹配到同样的网络区域,他们对于网络注释的贡献,会部分冗余。为了把这种冗余最小化并简化注释过程,基于他们在富集地图中的相似性,SAFE把这些terms归到一个组。产生的19个区域,用不同的颜色来代表,并且用tag lists来标注,形成一个综合性的,系统的和定量的基于GO的功能富集地图,也就是GIS网络。
由SAFE产生的功能地图,和手工注释的网络高度一致。尤其,SAFE鉴定了所有的手工注释的区域,并且和GO条目联系在一起,匹配手工指定的标签。值得注意的是,SAFE也识别了三个之前没有注释的网络区域,这种丢失或许因为他们的size小和特异性定位。
Analysis of Robustness
为了证实第一个假设,我验证了假如node距离被定义为map-independent 度量,比如,相关系数权重的最短路径长度correlation-weighted shortest path length(CSPL).在CSPL中,每一个边的权重是1-R, R是两个链接的genes的遗传相互作用profiles,并且这个value没有参与网络的构建。由MSPL和CSPL生成的富集全景图之间的median相似性为密度=0.7(斯皮尔曼rank相关系数)2f。这显示,无论这个距离度量是什么,大多数的邻居保持富集于同一个GOterms到一个相似的degree,并且MSPL依赖于估算node距离。
默认情况下,所有网络距离中在最低的5%的在彼此的邻居里。我验证了这个假设影响邻居富集的程度,通过测验设定一个更大的距离阈值。我发现,增加的或减少的默认阈值,多到2倍的产生高度相似的富集landscapes(median 密度=0.78和0.81,相应的fig2g),这对富集的GOterms有以一个有限的影响。这显示了,邻居富集对邻居的size不敏感,并且距离阈值的选择在2倍范围内变化不重要。
最后我评估了网络富集对spring-embedded layout algorithm的非决定性本质的稳健性,这种算法移除了所有的nodes(这些nodes在每一次run中都有一个平衡的位置)。通过重复的应用这个layout并比较产生的网络地图的富集全景,我发现,平均而言,任何两个独立的maps的富集全景都高度相似(median across all GO 条目密度=0.82,),这显示,不管跨越layout runs的绝对的节点位置的不同,邻居保持大部分不变。为了消除残余的可变性,以确保完整的可重复性,SAFE可以被设置来控制layout算法的随机step,并且在每次run中产生同样的node位置。
网络注释的精确性或许也依赖于功能注释标准的质量。为了验证SAFE对注释噪音的稳健性,我系统的通过随机引入不同数目的假阳性或假阴性注释来改变了所有的GO bp terms,并且比较产生的富集全景图fig2I。没啥问题
SAFE促进功能基因集的整合
因为所有富集标准的内在的偏见和局限,用单一类型的生物信息比如GO对一个网络进行注释,不可能产生网络功能组织的全景地图。一个更有效的策略是使用多个功能数据的独立资源,并且迭代的应用他们来注释同一个网络。这样的方法不进可以产生关于网络的更真实的描述也可以揭示data 类型之间的始料未及的联系。
在酵母中,化学基因组学产生了功能信息的丰富的资源。在一个化学基因组screen中,基因组范围的酵母突变体的数据在增长,(在多种化学复合物),并且,每一种突变都有未处理的哦。鉴定对给定的化学物敏感或不敏感的突变体对mapping 通路非常重要,而这个通路介导这个复合物的毒性或对保护细胞免于致死效应是必须的。我假说推断SAFE通过用化学基因组data注释GIS网络可以协助鉴定这些通路,并且可以鉴定对化学物敏感或不敏感的功能网络区域。
为了验证这个假设,我使用最近的一个化学基因组数据,它测量了大量的fittness分数,对大概5000个酵母同源删除突变体,暴露于132个化学复合物,这些化合物有已知的作用模式。使用这些数据,SAFE产生了132个复合物特异的fitness富集全景图,并且mapped相应的敏感和抵抗的突变体,通过GIS网络,fig3ABC.
通过对fitness富集全景图的分析(GO biological process),显示,全景图与我们当前的知识高度一致。例如,对doxorubicin这个DNA嵌入剂敏感的突变体,通过阻断拓扑异构酶II来阻断DNA复制,这种突变体主要在以下生理活动相关的网络region中富集,DNA复制和修复,相似的,一个靶向线粒体的蛋白质合成抑制剂在核糖体和线粒体相关的GO条目也富集重叠。
Case study:SAFE揭示抗Bortezomib(蛋白酶体抑制剂硼替佐米)的机制
除了已知的复合物作用模式,SAFE也可以揭示新的反应模式。
一个例子是关于bortezomib的fitness富集全景图,这是一种蛋白酶体抑制剂,已经证实可以用了治疗多发性骨髓病,多发性淋巴瘤和其它几种正在接收临床实验的类型的癌症。SAFE分析显示,在酵母中,对bortezomib敏感的突变体主要在和蛋白酶体介导的蛋白降解,细胞周期控制和转录调控相关网络区域富集fig3c。这些结果与人类细胞中的发现一致,bortezomib不直接通过保护促死亡因子来促进程序化细胞死亡,是协同的组蛋白去甲基化抑制剂,来调控转录。
值得注意的是,SAFE也显示,抗bortezomib和网络中的分泌和囊泡介导的转运go term强烈相关fig3c。虽然几个报道已经显示,蛋白酶体的失活可以通过内质网中的错误折叠蛋白的聚集引起内质网stress,但是没有预料失去功能的突变体在ER或其他囊泡相关的功能可以减弱这个或其他蛋白媒体相关的压力。有一个直接的关于bortezomib的实验可以证实SAFE的富集分析:对药物最强抵抗的四个突变株,执行了完全或部分的敲除在YTP6,RIC1,RGP1基因,调控形成,移动或囊泡融合,从高尔基体区域。另外,几个卷入高尔基相关的其他价格i额蛋白也在抗突变的前15之内。
这个发现的统计学重要性也被网络独立基因集GSEA支持。GSEA决定功能组的成员是否倾向于在ranked基因列表的top或底部出现,然后来检测这种分布的可能性。通过对ranked list of bortezomib fitness得分应用GSEA,所有4373个GO BP,我确认了想高尔基体内囊泡介导的转运和细胞质到囊泡靶向途径显著富集。和SAFE的结果相比,然而,这些pathways的首要的在GSEA中却更少的出现:
GSEA检测到的48个显著的GO条目的大多数(58%)涉及离子稳态,细胞内pH调节,还有其他距离相关的功能相关的。这种不一致显示了,通过影响网络拓扑性,SAFE可以检测功能信号,这些信号在网络非依赖型分析中不容易出现。
更复杂网络的注释:包括蛋白蛋白相互作用网络
和其他生物网络相比,GIS网络相对稀少和模块化fig2a,并且也可能顺从于注释。为了评估是否SAFE也可以用来注释更复杂的网络,我首先验证了它是否可以在一个GIS网络更稠密的版本中检测到功能富集信息,这个版本通过降低GIS阈值的最小值来获取,这个最小值是连接性所需要的。对GIS网络的注释with40%-240%更多的边比原始产生的相似性GO条目富集全景图(median 密度=0.61-0.75),显示不管网络的密度,SAFE对内在的功能结构比较敏感。
也可能,更复杂的生物网络没有一个像GIS一样重要的结构,这样注释更加困难。因此,我广泛的PPI 网络,在5699个酵母蛋白质中产生了多至78406个物理绑定。PPI优先链接相同的蛋白复合体成员和其他功能相关的蛋白,这个网络地图,在全局范围内没有显示出可见的拓扑结构fig4a。然而,SAFE揭示了网络中的21个大的功能域,每个domain和一个明确的GO term富集,还有一个唯一的富集的go terms列表fie4B。这个富集地图结果强烈显示了物理结合,就像GIS,可以把蛋白质聚成一个大的功能单位,这个大的功能单位超越了蛋白复合体和分子通路。这些复合体的相对网络定位,连同他们的GIS部分,对酵母细胞的功能组织或许可以提供一个新的视野。
DISCUSSION
这里,我描述了SAFE的发展,验证和应用,这是一种注释生物网络,检测其功能组织的自动化方法。考虑到网络和可视化map的连接性,SAFE定位所有的网络区域,在一个或更多功能属性富集的,比如GOterm,或定量表型。这个过程中,SAFE回答三个基本的问题,
第一,网络中的任何区域是不是特异性的和一个给定的功能或表型相关?
第二,这些区域定位在网络的什么位置
第三,他们的定位如何与其他功能或表型进行比较
通过回答这些问题,SAFE建立了一个网络功能地图,并探索细胞内的过程关系。
通过可视化的力量,SAFE也可以用来提高我们对功能标准的理解。例如,SAFE 用GO生物过程进行的GIS网络的注释显示,一些GO term富集在单一的网络区域,而其他的是多区域。虽然多区域GO terms比特定区域的数目要大,但是他们的size分布会产生更多的重叠。这显示了term size不仅仅贡献于全景差异。一个有趣的可能是,区域特异的GO term共享一个功能特异的相似性水平,这是有GIS网络的拓扑型定义的。如果事实如此,这些terms可以用来delineate描绘一个GO等级聚类的剖面(cross-section)并且产生GO 注释的扁平subset,和GO slim相似。在基因组中扁平化注释很重要,这归于它们的小size和低的冗余。SAFE或许可以提供产生数据驱动的网络特异性注释标准的生成,这可以使得更多的靶向的功能分析成为可能并简化他们的解释。
SAFE功能maps的解释会从一个更好的网络layout算法中受益。数据驱动的网络输出,比如spring embedded, 是一种非监督方法可以基于nodes的连接度来组织nodes,并且事实上是多维度还原程序。在这个默认setup中,SAFE依赖layouts鉴定本地邻居并且匹配他们的功能富集。然而,一个特别的layout如何被选择,我们仍然知道的太少。不管他们在揭示数据内部的隐含模式的巨大潜能,layouts可以典型用来产生esthetically pleasing(赏心悦目的) network visualizations(赏心悦目的网络可视化)全景图并且很少是任何系统网络分析的基础。结果而言,我们在评估网络输出方面有有限的经验,并且在不同的网络中相对的表现有很少的理解。SAFE或许对同一个网络使用普通的功能属性进行可变的layouts有一些评估作用。理想状态是,这样的分析能够为每一个网络类型鉴定最佳的layouts并且为比较网络建立共同的ground。
生物网络的定量比较是系统生物学的大目标。对不同网络类型中的genes,通路和过程是如何连接的深入理解有助于对开发成功的策略(对一个细胞中,整合多个网络成一个single综合的模型)。通过把相同的功能属性map到不同的网络富集中,SAFE可以对这个目标有重要的贡献。然而,谨慎的统计学方法一定被内置来比较SAFE的跨网络maps并且得出有意义的结论,关于他们的不同和相似。
总之,SAFE对网络中的功能组织提供了一个总体的视角,通过在功能groups和网络区域之间map统计学联系。和其他网络分析的方法相比(其他方法抽取网络区域并独立比较他们)。SAFE显示,网络输出合并它们的稳健富集分析,是一个有效的分析完整分子网络的策略,可以对他们代表的生物系统获得视角。
Natrue biotechnology文章
Dosage suppression genetic interaction networks enhance functional wiring diagrams of the cell
DOI: 10.1038/nbt.1855 · Source: PubMed
Results
A global dosage suppression genetic interaction network
我们收集了424个必须基因的一系列dosage suppression genetic interactions ,我们称之为query基因,他们在SGD中被注释。这些相互作用形成一个网络包含768个genes和1293条边。大部分query genes只有少数dosage suppressors,虽然少数基因有大的interactions。我们在cytoscape中使用force-directed layout展示,这样的,拥有共同的dosage suppression 相互作用的genes会形成清晰的明显的clusters。Markov聚类分析鉴定出9个clusters,每个包含大于等于30个genens,都对应特定的生物学过程。和综合的基因网络相似,这些clusters之间的相对距离看起来反应了共享的或共有的功能。(大概意思是说,网络中离的近,说明功能有相似性,基因有重叠)例如,下图中vesicle-mediated transport,exocytosis和细胞生长和形态生成三个cluster的genes在网络的相对接近揭示了他们之间的功能联系,这揭示了,这种相互作用可以独立的用来cluster基因,基于功能相互关系。