- 数学推理中在推理规模化下检查假阳性解
硅谷秋水
大模型机器学习人工智能语言模型深度学习机器学习人工智能
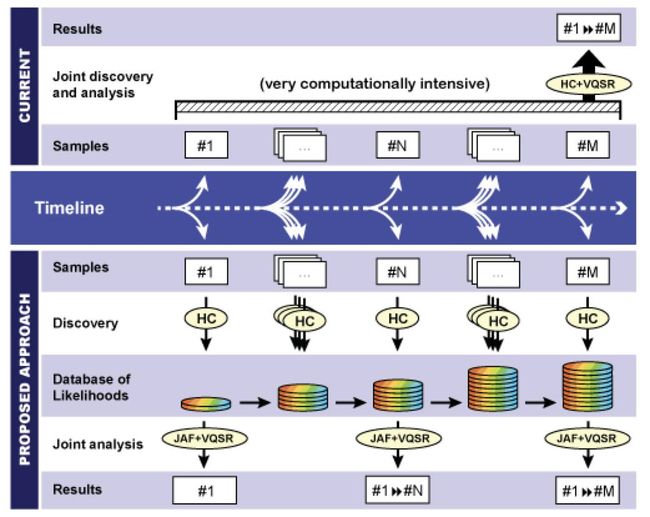
25年2月来自中科大和微软亚洲研究院的论文“ExaminingFalsePositivesunderInferenceScalingforMathematicalReasoning”。语言模型的最新进展已带来各种基准测试中数学推理能力的显著提升。然而,大多数基准测试依赖于自动评估方法,这些方法仅使用启发式方法比较最终答案,而不验证底层推理步骤。这种限制导致假阳性解,其中模型可能会产生正确的最终答案
- 清华大学第四发《DeepSeek+DeepResearch 让科研像聊天一样简单》
人工智能
当下科研领域,传统模式急需改变,清华大学第四版《DeepSeek+DeepResearch:让科研像聊天一样简单》全文一共86页,以下是文档的关键内容总结:一、智能组合优势DeepSeek与DeepResearch构建先进技术体系,有强大模型运算、智能数据处理和友好交互界面。模型在数据处理速度、精准度和泛化能力上远超传统模型。数据采集渠道广、处理快,能读取多种格式文件。数据分析深入,可视化直观,还
- 【OpenTiny调研征集】共创技术未来,分享您的声音!
前端vue.js开源
欢迎参与2025年OpenTiny开源社区用户调研征集调研背景随着OpenTiny开源项目的不断发展,我们一直致力于为开发者提供高质量的Web前端开发解决方案。为了更好地满足用户需求,提升项目的实用性和易用性,我们决定发起一项用户调研活动,诚挚邀请您参与。调研目的了解用户需求:收集您在使用OpenTiny开源项目过程中的需求、问题和建议,以便我们更好地改进和优化。提升用户体验:通过您的反馈,我们将
- 基于微信小程序的宠物寄养平台的设计与实现
图灵软件设计
JAVASSM小程序微信小程序小程序springbootmaven后端javamybatis
现在宠物寄养管理中已有一些商家使用了基本的管理软件,这些软件都是依靠客户端,只可以特定人员使用,不能实现信息的共享。虽然可以帮助工作人员减少工作量,但从根本上还是无法满足用户的需求。这些软件都还是基于网络发展之初的要求,没有利用现代网络的技术,体现不了更为实用的功能。依靠客户端的系统开发时没有考虑园际化的问题,所以也满足不了国际化的要求。最近几年来,我国网络快速发展,传统的管理方式也越来越适应不了
- mysql实时同步到es
数据库
测试了多个方案同步,最终选择oceanu产品,底层基于Flinkcdc1、实时性能够保证,binlog量很大时也不产生延迟2、配置SQL即可完成,操作上简单下面示例mysql的100张分表实时同步到es,优化备注等文本字段的like查询创建SQL作业CREATETABLEfrom_mysql(idint,cidintNOTNULL,gidbigintNOTNULL,contentvarchar,c
- Python学习心得两大编程思想
lifegoesonwjl
python开发语言pycharm前端c语言
一、两大编程思想:1.面向过程:功能上的封装典型代表:C语言2.面向对象:属性和行为上的封装典型代表:Python、Java二、面向过程与面向对象的异同点:1.区别:面向过程:事物比较简单,可用线性的思维去解决面向对象:事务比较复杂,使用简单的线性思维无法解决2.共同点:(1)面向过程和面向对象都是解决实际问题的一种思维方式;(2)二者相辅相成,并不是对立的;(3)解决复杂问题,通过面向对象方式便
- Linux升级Anacodna并配置jupyterLab
伪_装
环境部署linux服务器Anacondapythonjupyter
在使用Anaconda的过程中,随着项目和需求的发展,可能需要升级Anaconda的Base环境中的Python版本。本文将详细介绍如何安全地进行升级,包括步骤、代码示例与最终流程图。升级Python一、环境准备在进行任何升级之前,建议先检查当前的Python版本以及各个库的兼容性。我们可以通过以下命令检查当前的Python版本:condainfo你会看到类似以下的输出,其中包含了当前Python
- 信息获取、扫描与服务识别、漏洞验证、嗅探攻击、代理与隧道、metasploit渗透攻击等
Utopia.️
web安全安全网络
1.信息获取信息获取是渗透测试和安全评估的第一步,主要目的是收集目标系统的各种信息。这些信息可以帮助确定攻击面和潜在的安全漏洞。技术和工具:域名信息:使用whois查询域名注册信息。DNS查询:使用nslookup或dig获取DNS记录,包括A记录、MX记录等。网络扫描:使用nmap或Masscan扫描目标网络,收集IP地址和开放端口信息。公开信息:通过搜索引擎、社交媒体、公司网站等公开资源获取目
- Nginx配置反向代理不成功的原因(Docker安装版)
程序员迪迦
项目实战nginxdockerlinux
问题背景在linux服务器中使用docker下载了Nginx,然后根据网上的教程来配置反向代理的时候发现80端口无法访问server块的配置server{listen80;server_name127.0.0.1;#access_log/var/log/nginx/host.access.logmain;location/{proxy_passhttp://127.0.0.1:8080;#inde
- Android arcgis加载在线底图
Angie洛林
androidarcgis
我整理的一些关于【信息系统】的项目学习资料(附讲解~~)和大家一起分享、学习一下:https://edu.51cto.com/mic-position/757.html在Android中使用ArcGIS加载在线底图ArcGIS是Esri提供的一套强大的地理信息系统(GIS)解决方案,支持多种平台,包括Android。本文将介绍如何在Android应用中使用ArcGIS加载在线底图,并配有相关代码示
- 使用Arcgis API for android加载OpenStreetMap底图并完成定位
续汉冕
移动开发androidandroidstudioarcgisapi
为了完成这个应用功能花了三天,代码倒不多就是比较坑!环境:AndroidSDKAPI22,AndroidStudio1.2.2,arcgisandroidSDK10.2.7如何基于ArcgisAPIforandroid在AndroidStudio新建一个项目就不再赘述了,大家可以参考以下网址:使用AndroidStudio与ArcgisandroidSDK的开发环境部署和HelloWorld:ht
- cesium(vue)一些面试问题(包含Three.js)
GIS瞧葩菜
vue.jsjavascriptcesium
1.在不同的应用场景和技术栈中,模型加载方法和格式有所不同,下面主要从Web前端三维场景(使用Three.js和cesium)使用Three.js加载模型常见模型格式及加载方法GLTF/GLB格式格式特点:GLTF(GraphicsLibraryTransmissionFormat)是一种开放的、基于JSON的三维模型传输格式,GLB是其二进制版本。它们具有文件小、加载快、支持动画、材质和骨骼等优
- sql拼接错误直到数据全部删除
数据库
起引订单表的扩展表,在配货转发货过程中会删除配货库数据后,插入到发货库。但一直有数据在没有转移的情况下也被删除。查找通过解析binlog和审计,最终查到DELETEFROMorder.order_extendWHERE1234开始以为sql审计有问题,后来发现该语句效果同where1=1,直接导致全表删除。解决使用binlog2sql回滚数据;修复sql拼接错误。
- 动态规划之背包问题全解
学会了,不,学废了
动态规划
概述———动态规划提出人:理查德·贝尔曼本质:一张表格处理方法内容:把原问题分解为若干子问题,自底向上先求解最小子问题,把结果储存在表格中,求解大的子问题时直接从表格中查询小的子问题的解,以避免重复计算,从而提高效率。一、动态规划求解原理适用范围:问题需要具备3个性质———最优子结构、子问题重叠、无后效性。最优子结构指问题最优解包含其子问题的最优解,是使用动态规划的基本条件。三要素:状态、阶段、决
- ArcGIS二次开发之WPF中控件的使用
ShirmyMao
ArcGIS二次开发wpfc#arcgis
WPF中ArcGIS控件的使用WPF中插入ArcGIS控件Winform控件上嵌套使用WPF控件WPF中插入ArcGIS控件在WPF中引用ArcGIS的控件需要使用WindowsFromsHost,具体用法如下:添加引用:WindowsFormsIntegration和system.windows.formWpf.xaml中后台代码中:publicAxMapControlMapControl=ne
- Jupyter使用Nginx做反向代理
syuszu
nginx
1.在/nginx/conf/nginx.conf上进行修改因为jupyter对于header有过滤,需要将header复制,实例如下:server{server_name127.0.0.1;#入口地址listen80;location/{proxy_passhttp://127.0.0.1:8888;#jupyter服务器地址proxy_set_headerHost$host;proxy_set
- java实现,使用向量相似度 输入字符串,在定义好的字符串集合中根据语义匹配出最准的一个。
melck
1024程序员节
以下是完整的Java示例代码,包括字符串集合的定义和根据输入字符串匹配最相似字符串的逻辑:importjava.util.*;publicclassSemanticMatching{publicstaticvoidmain(String[]args){//定义字符串集合ListstringCollection=Arrays.asList("Whereistherestroom?","Canyout
- java 实现TextRank算法提取文章摘要
melck
java算法开发语言
在Java中,常用的文章摘要提取库是“TextRank”算法。该算法从文本中提取主题和段落,并根据主题和文本中的单词计算权重。使用TextRank实现文章摘要提取具体步骤如下:寻找文章中的关键句子:首先需要分割出文章中的句子,可以使用分词库将文章拆分成句子,然后使用TextRank算法找到文章中与主题相关的句子,这些句子通常包含有标题、关键字等。计算句子的权重:针对关键句子,需要对每个句子计算权重
- AI 如何接口调试?可以展示推理过程
人工智能深度学习机器学习
如何在开发AI接口的同时,能看到实时的AI回复,避免传统的轮询方式,而无需长时间等待。常用的AI模型(比如Deepseek、Gemini)都是支持流式输出,那有没有一款API接口软件可以实现这功能?近期Apifox增强了调试SSE接口功能,实现了发起HTTP请求流式响应就会自动合并为可读文本,实时以自然语言呈现响应。而且针对Deepseek还能展示思考推理过程!这大大降低AI应用开发难度,有图为证
- Win7因为更改了硬件或软件导致无法开机怎么解决?
m0_70960708
笔记服务器运维
如果是因为硬件导致的无法开机或者是因为软件的问题导致无法开机的问题该怎么解决呢?其实这个问题的原因一般是因为用户最近更改了硬件或者软件,导致电脑开机出现问题,所以针对这个问题有特定的解决办法,一起来看看如何解决吧。方法一:恢复上一次的设置1、首先我们使用电源键重启电脑,接着在开机时按下“F8”进入高级系统选项。方法二:删除驱动1、如果还是开不了机,再重启电脑按“F8”,然后选择进入安全模式,如图所
- 萌新的51之旅——串口通信(3)
codoger
单片机
一,过程特性过程特性规定了信号之间的时序关系,以便正确的接收和发送数据采用RS-232c接口存在的问题一,传输距离短,传输速率低该总线标准受电容允许值的约束,使用时传输距离一般不要超过15米,最高传输速率为20K二,有电平偏移该总线标准要求收发双方共地通信,距离较大时,收发双方的地电位差别较大,在信号地上将有比较大的地电流,并产生压降三,抗干扰能力差该接口的电瓶转换时采用单端输入输出,在传输过程中
- 2020-12-24 CH340使用注意事项
billgodark
笔记
留存谨记!,CH340绑定封装RS232接口芯片的功耗较大,TX和RX电流可能拉低电平,在实际使用时需要在Tx和Rx上串行470Ω左右的电阻,绑定版CH340的USB转RS232电平串行口建议使用这种方式
- OpenAI 深度研究与 Gemini 深度研究:哪个更好?
知识小报童
DeepSeek人工智能深度学习机器学习神经网络自然语言处理语言模型AIGC
目录*什么是OpenAI深度研究?**OpenAI深度研究的关键特性:**OpenAI深度研究的应用:**使用案例:**什么是Gemini深度研究?**Gemini深度研究的关键特性:**Gemini深度研究的应用:**使用案例:**Gemini2.0Flash:增强性能**可用性:**OpenAI深度研究与Gemini深度研究:详细比较**OpenAI深度研究与Gemini深度研究之间的关键区别
- 总结10个Python赚钱的接单平台 兼职月入5000+
begefefsef
面试学习路线阿里巴巴android前端后端
前言“如果说当下什么编程语言最靠谱或者比较适合搞副业?”答案肯定100%是:Pythonpython是所有语法中最简单易上手的语言,不需要特别的的英语词汇量,逻辑思维也不需要很差就能上手。而且学会了之后就能编写代码爬取各种数据,制作各种图表,提升工作效率。而且还能利用业余时间接点私活,一个月轻松收入过万不是问题,这样的生活他不香吗?今天就给大家盘点几个基本入门接私活的资源,让你轻松学python,
- 同步盘怎么选?2025年这三款网盘的功能和优缺点全在这!
SJ_HP
经验分享远程工作安全百度云
在数字时代,文件存储、多设备同步和团队协作已经成为我们生活和工作中不可或缺的一部分。无论是个人用户还是企业团队,都面临着文件存储空间不足、设备间同步困难以及团队合作效率低下的痛点。同步盘和企业云盘的出现,正是为了解决这些问题。它们不仅提供了便捷的文件存储和管理功能,还通过多设备同步和团队协作功能,极大地提升了工作效率。今天,我们将对比几款热门的同步盘产品,帮助你找到最适合自己的解决方案。亿方云:企
- USB转串口芯片CH9102替代CP2102注意事项
Chery1140
单片机嵌入式硬件
CH9102与CP2102可实现pin2pin兼容,可以在不更改硬件设计的前提下实现不同型号间快速切换与产品应用。CH9102系列型号包括:CH9102F(QFN24)和CH9102X(QFN28),CP2102系列型号包括:CP2102、CP2102N-GQFN24、CP2102N-GQFN28。1.应用差异说明1)驱动说明:CH9102芯片为CDC类串口芯片,用户可以选择使用操作系统内置的CD
- 【计算机毕设任务书】基于微信小程序的宠物寄养平台的设计与实现
Eastonzhang888
计算机毕设任务书参考案例课程设计微信小程序宠物数据库intellij-idea计算机毕业设计小程序
一、设计的主要内容、技术参数及工作要求研究目的现在宠物寄养管理中已有一些商家使用了基本的管理软件,这些软件都是依靠客户端,只可以特定人员使用,不能实现信息的共享。虽然可以帮助工作人员减少工作量,但从根本上还是无法满足用户的需求。这些软件都还是基于网络发展之初的要求,没有利用现代网络的技术,体现不了更为实用的功能。依靠客户端的系统开发时没有考虑园际化的问题,所以也满足不了国际化的要求。最近几年来,我
- EasyX安装及使用
于冬恋
java开发语言
安装链接:EasyXGraphicsLibraryforC++安装完成包含头文件graphics.h即可使用RGB合成颜色(红色部分,绿色部分,蓝色部分)每种颜色的值都是(0~255)坐标默认的原点在窗口的左上角,x轴向右为正,y轴向下为正,度量单位是像素点。设备:简单来说就是绘图表面(在EasyX中,设备分为两种,一种是默认的绘图窗口,一种是IMAGE对象。通过SetWorkinglmage()
- AI 大模型:Intelligent Agent—— 开启智能新纪元
AI-入门
人工智能学习产品经理面试agi
在LLM语境下,Agent理解为在某种能自主理解、规划决策、执行复杂任务的智能体,LLM充当着智能体的“大脑”。从软件工程的角度,智能体是一种基于大语言模型的,具备规划思考能力、记忆能力、使用工具函数的能力,能自主完成给定任务的计算机程序。在基于LLM的智能体中,LLM的充当着智能体的“大脑”的角色,同时还有3个关键部分:规划(Planning):智能体会把大型任务分解为子任务,并规划执行任务的流
- Anaconda 环境克隆、迁移 ,用Anaconda里面的conda命令创建虚拟环境并克隆环境或者复旧电脑实验环境包、_conda复制环境
好像要长脑子了1
程序员conda
###9、设置国内镜像http://Anaconda.org的服务器在国外,安装多个packages时,conda下载的速度经常很慢。清华TUNA镜像源有Anaconda仓库的镜像,将其加入conda的配置即可:#添加Anaconda的TUNA镜像condaconfig--addchannelshttps://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/f
- C/C++Win32编程基础详解视频下载
择善Zach
编程C++Win32
课题视频:C/C++Win32编程基础详解
视频知识:win32窗口的创建
windows事件机制
主讲:择善Uncle老师
学习交流群:386620625
验证码:625
--
- Guava Cache使用笔记
bylijinnan
javaguavacache
1.Guava Cache的get/getIfPresent方法当参数为null时会抛空指针异常
我刚开始使用时还以为Guava Cache跟HashMap一样,get(null)返回null。
实际上Guava整体设计思想就是拒绝null的,很多地方都会执行com.google.common.base.Preconditions.checkNotNull的检查。
2.Guava
- 解决ora-01652无法通过128(在temp表空间中)
0624chenhong
oracle
解决ora-01652无法通过128(在temp表空间中)扩展temp段的过程
一个sql语句后,大约花了10分钟,好不容易有一个结果,但是报了一个ora-01652错误,查阅了oracle的错误代码说明:意思是指temp表空间无法自动扩展temp段。这种问题一般有两种原因:一是临时表空间空间太小,二是不能自动扩展。
分析过程:
既然是temp表空间有问题,那当
- Struct在jsp标签
不懂事的小屁孩
struct
非UI标签介绍:
控制类标签:
1:程序流程控制标签 if elseif else
<s:if test="isUsed">
<span class="label label-success">True</span>
</
- 按对象属性排序
换个号韩国红果果
JavaScript对象排序
利用JavaScript进行对象排序,根据用户的年龄排序展示
<script>
var bob={
name;bob,
age:30
}
var peter={
name;peter,
age:30
}
var amy={
name;amy,
age:24
}
var mike={
name;mike,
age:29
}
var john={
- 大数据分析让个性化的客户体验不再遥远
蓝儿唯美
数据分析
顾客通过多种渠道制造大量数据,企业则热衷于利用这些信息来实现更为个性化的体验。
分析公司Gartner表示,高级分析会成为客户服务的关键,但是大数据分析的采用目前仅局限于不到一成的企业。 挑战在于企业还在努力适应结构化数据,疲于根据自身的客户关系管理(CRM)系统部署有效的分析框架,以及集成不同的内外部信息源。
然而,面对顾客通过数字技术参与而产生的快速变化的信息,企业需要及时作出反应。要想实
- java笔记4
a-john
java
操作符
1,使用java操作符
操作符接受一个或多个参数,并生成一个新值。参数的形式与普通的方法调用不用,但是效果是相同的。加号和一元的正号(+)、减号和一元的负号(-)、乘号(*)、除号(/)以及赋值号(=)的用法与其他编程语言类似。
操作符作用于操作数,生成一个新值。另外,有些操作符可能会改变操作数自身的
- 从裸机编程到嵌入式Linux编程思想的转变------分而治之:驱动和应用程序
aijuans
嵌入式学习
笔者学习嵌入式Linux也有一段时间了,很奇怪的是很多书讲驱动编程方面的知识,也有很多书将ARM9方面的知识,但是从以前51形式的(对寄存器直接操作,初始化芯片的功能模块)编程方法,和思维模式,变换为基于Linux操作系统编程,讲这个思想转变的书几乎没有,让初学者走了很多弯路,撞了很多难墙。
笔者因此写上自己的学习心得,希望能给和我一样转变
- 在springmvc中解决FastJson循环引用的问题
asialee
循环引用fastjson
我们先来看一个例子:
package com.elong.bms;
import java.io.OutputStream;
import java.util.HashMap;
import java.util.Map;
import co
- ArrayAdapter和SimpleAdapter技术总结
百合不是茶
androidSimpleAdapterArrayAdapter高级组件基础
ArrayAdapter比较简单,但它只能用于显示文字。而SimpleAdapter则有很强的扩展性,可以自定义出各种效果
ArrayAdapter;的数据可以是数组或者是队列
// 获得下拉框对象
AutoCompleteTextView textview = (AutoCompleteTextView) this
- 九封信
bijian1013
人生励志
有时候,莫名的心情不好,不想和任何人说话,只想一个人静静的发呆。有时候,想一个人躲起来脆弱,不愿别人看到自己的伤口。有时候,走过熟悉的街角,看到熟悉的背影,突然想起一个人的脸。有时候,发现自己一夜之间就长大了。 2014,写给人
- Linux下安装MySQL Web 管理工具phpMyAdmin
sunjing
PHPInstallphpMyAdmin
PHP http://php.net/
phpMyAdmin http://www.phpmyadmin.net
Error compiling PHP on CentOS x64
一、安装Apache
请参阅http://billben.iteye.com/admin/blogs/1985244
二、安装依赖包
sudo yum install gd
- 分布式系统理论
bit1129
分布式
FLP
One famous theory in distributed computing, known as FLP after the authors Fischer, Lynch, and Patterson, proved that in a distributed system with asynchronous communication and process crashes,
- ssh2整合(spring+struts2+hibernate)-附源码
白糖_
eclipsespringHibernatemysql项目管理
最近抽空又整理了一套ssh2框架,主要使用的技术如下:
spring做容器,管理了三层(dao,service,actioin)的对象
struts2实现与页面交互(MVC),自己做了一个异常拦截器,能拦截Action层抛出的异常
hibernate与数据库交互
BoneCp数据库连接池,据说比其它数据库连接池快20倍,仅仅是据说
MySql数据库
项目用eclipse
- treetable bug记录
braveCS
table
// 插入子节点删除再插入时不能正常显示。修改:
//不知改后有没有错,先做个备忘
Tree.prototype.removeNode = function(node) {
// Recursively remove all descendants of +node+
this.unloadBranch(node);
// Remove
- 编程之美-电话号码对应英语单词
bylijinnan
java算法编程之美
import java.util.Arrays;
public class NumberToWord {
/**
* 编程之美 电话号码对应英语单词
* 题目:
* 手机上的拨号盘,每个数字都对应一些字母,比如2对应ABC,3对应DEF.........,8对应TUV,9对应WXYZ,
* 要求对一段数字,输出其代表的所有可能的字母组合
- jquery ajax读书笔记
chengxuyuancsdn
jQuery ajax
1、jsp页面
<%@ page language="java" import="java.util.*" pageEncoding="GBK"%>
<%
String path = request.getContextPath();
String basePath = request.getScheme()
- JWFD工作流拓扑结构解析伪码描述算法
comsci
数据结构算法工作活动J#
对工作流拓扑结构解析感兴趣的朋友可以下载附件,或者下载JWFD的全部代码进行分析
/* 流程图拓扑结构解析伪码描述算法
public java.util.ArrayList DFS(String graphid, String stepid, int j)
- oracle I/O 从属进程
daizj
oracle
I/O 从属进程
I/O从属进程用于为不支持异步I/O的系统或设备模拟异步I/O.例如,磁带设备(相当慢)就不支持异步I/O.通过使用I/O 从属进程,可以让磁带机模仿通常只为磁盘驱动器提供的功能。就好像支持真正的异步I/O 一样,写设备的进程(调用者)会收集大量数据,并交由写入器写出。数据成功地写出时,写入器(此时写入器是I/O 从属进程,而不是操作系统)会通知原来的调用者,调用者则会
- 高级排序:希尔排序
dieslrae
希尔排序
public void shellSort(int[] array){
int limit = 1;
int temp;
int index;
while(limit <= array.length/3){
limit = limit * 3 + 1;
- 初二下学期难记忆单词
dcj3sjt126com
englishword
kitchen 厨房
cupboard 厨柜
salt 盐
sugar 糖
oil 油
fork 叉;餐叉
spoon 匙;调羹
chopsticks 筷子
cabbage 卷心菜;洋白菜
soup 汤
Italian 意大利的
Indian 印度的
workplace 工作场所
even 甚至;更
Italy 意大利
laugh 笑
m
- Go语言使用MySQL数据库进行增删改查
dcj3sjt126com
mysql
目前Internet上流行的网站构架方式是LAMP,其中的M即MySQL, 作为数据库,MySQL以免费、开源、使用方便为优势成为了很多Web开发的后端数据库存储引擎。MySQL驱动Go中支持MySQL的驱动目前比较多,有如下几种,有些是支持database/sql标准,而有些是采用了自己的实现接口,常用的有如下几种:
http://code.google.c...o-mysql-dri
- git命令
shuizhaosi888
git
---------------设置全局用户名:
git config --global user.name "HanShuliang" //设置用户名
git config --global user.email "
[email protected]" //设置邮箱
---------------查看环境配置
git config --li
- qemu-kvm 网络 nat模式 (四)
haoningabc
kvmqemu
qemu-ifup-NAT
#!/bin/bash
BRIDGE=virbr0
NETWORK=192.168.122.0
GATEWAY=192.168.122.1
NETMASK=255.255.255.0
DHCPRANGE=192.168.122.2,192.168.122.254
TFTPROOT=
BOOTP=
function check_bridge()
- 不要让未来的你,讨厌现在的自己
jingjing0907
生活 奋斗 工作 梦想
故事one
23岁,他大学毕业,放弃了父母安排的稳定工作,独闯京城,在家小公司混个小职位,工作还算顺手,月薪三千,混了混,混走了一年的光阴。 24岁,有了女朋友,从二环12人的集体宿舍搬到香山民居,一间平房,二人世界,爱爱爱。偶然约三朋四友,打扑克搓麻将,日子快乐似神仙; 25岁,出了几次差,调了两次岗,薪水涨了不过百,生猛狂飙的物价让现实血淋淋,无力为心爱银儿购件大牌
- 枚举类型详解
一路欢笑一路走
enum枚举详解enumsetenumMap
枚举类型详解
一.Enum详解
1.1枚举类型的介绍
JDK1.5加入了一个全新的类型的”类”—枚举类型,为此JDK1.5引入了一个新的关键字enum,我们可以这样定义一个枚举类型。
Demo:一个最简单的枚举类
public enum ColorType {
RED
- 第11章 动画效果(上)
onestopweb
动画
index.html
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/
- Eclipse中jsp、js文件编辑时,卡死现象解决汇总
ljf_home
eclipsejsp卡死js卡死
使用Eclipse编辑jsp、js文件时,经常出现卡死现象,在网上百度了N次,经过N次优化调整后,卡死现象逐步好转,具体那个方法起到作用,不太好讲。将所有用过的方法罗列如下:
1、取消验证
windows–>perferences–>validation
把 除了manual 下面的全部点掉,build下只留 classpath dependency Valida
- MySQL编程中的6个重要的实用技巧
tomcat_oracle
mysql
每一行命令都是用分号(;)作为结束
对于MySQL,第一件你必须牢记的是它的每一行命令都是用分号(;)作为结束的,但当一行MySQL被插入在PHP代码中时,最好把后面的分号省略掉,例如:
mysql_query("INSERT INTO tablename(first_name,last_name)VALUES('$first_name',$last_name')");
- zoj 3820 Building Fire Stations(二分+bfs)
阿尔萨斯
Build
题目链接:zoj 3820 Building Fire Stations
题目大意:给定一棵树,选取两个建立加油站,问说所有点距离加油站距离的最大值的最小值是多少,并且任意输出一种建立加油站的方式。
解题思路:二分距离判断,判断函数的复杂度是o(n),这样的复杂度应该是o(nlogn),即使常数系数偏大,但是居然跑了4.5s,也是醉了。 判断函数里面做了3次bfs,但是每次bfs节点最多