我们知道,16S rRNA基因普遍存在于细菌细胞,在细菌基因组中位于核糖体小亚基(约1540 bp),该区域兼顾保守性和高变性,含有10 个保守区域(Conserved Regions)和 9 个高变区域(variable Regions),保守区可用于设计引物进行目的片段的扩增,而通过对高变区的分析可以辨别细菌种类。因此,16S rRNA基因被认为是最适于细菌系统发育学研究和物种分类鉴定。目前用于16S rRNA基因深度测序的区域主要有V4区,V3-V4区、和V4-V5区等。
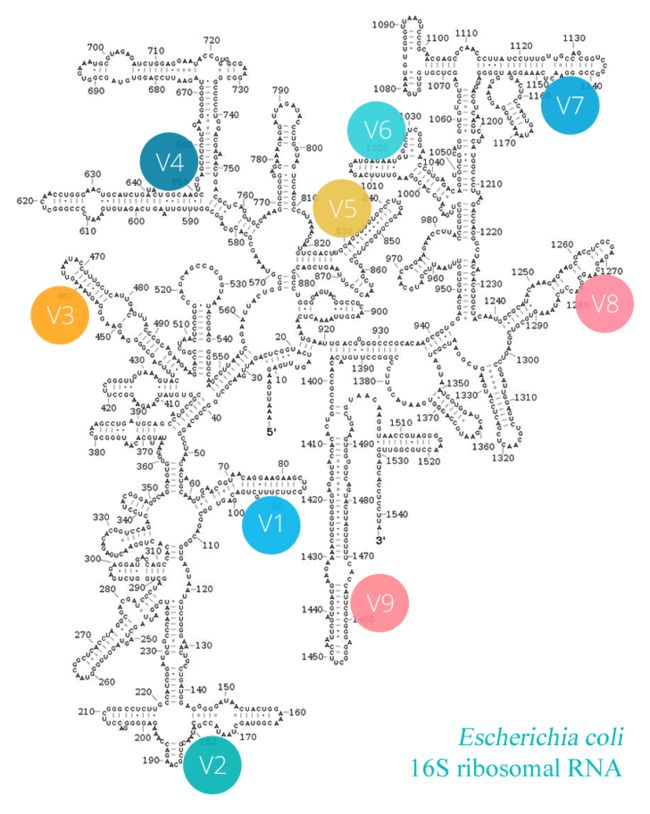
(大肠杆菌全长16S rDNA)
以Illumina Hiseq、Miseq为代表的第二代高通量测序平台的发展,已成为16S rDNA细菌多样性研究的主流手段。
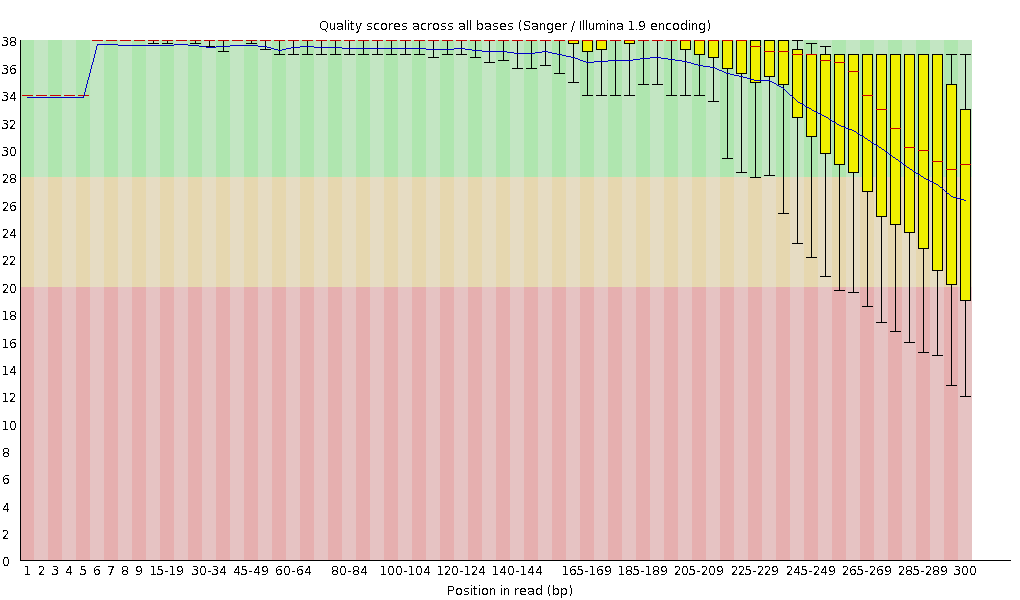
但是,由于Illumina测序策略本身的问题,导致其测序长度不可能太长。如下图,使用fastqc获得的raw read质量图,可以看到随着长度的延长,测序质量不断下降。
为什么Illumina测序读长短?
1. 在边合成边测序过程中,每个循环应该合成一个碱基,因为某些原因,例如酶活性的降低,导致一个循环没有合成碱基,这种少合成碱基的情况就称为Phasing。
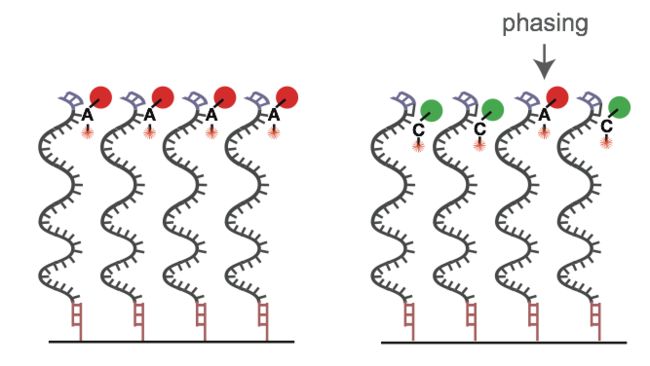
2. 同理,终止基团的脱落,导致一个循环合成二个或更多的碱基,这种情况称为Prephasing;
随着测序长度不断延长,不同步的信号越来越多,最后我们判断不出来到底是什么碱基被合成。此外,桥式PCR对文库长度的限制,以及激光对DNA链的打断也是读长的限制因素。
三代SMRT测序
单分子实时测序技术也被称为第三代测序技术(Thrid-generation sequencing,TGS),包括Oxford Nanopore的纳米孔测序技术和Pacific Biosciences单分子实时测序技术(Single Molecule Real-Time,SMRT)等,其中,纳米孔测序技术受限于其测序错误过高的问题,在16S rRNA基因测序等领域应用较少。目前,PacBio测序已被大量商用。
以Pacbio Sequel为代表的第三代单分子实时测序技术,应用于宏基因组学研究的优势除了长读长以外,还有由于建库过程中不需要PCR,弥补了GC-rich区域无法扩增的问题。
SMRT测序目前平均读长达>10kb,测序速度很快,每秒约10个dNTP。三代测序虽然测序错误率较高,但它的出错是随机的,并不会像第二代测序技术那样存在较多系统的测序错误的偏向,因而可以通过多次测序来进行有效的纠错。目前Sequel系统已能达到相当于Q50以上质量的测序准确度。
SMRT测序原理
PacBio SMRT的单分子测序和超长读长是如何实现的?我们重点看一下该技术的两点关键创新:分别是零模波导孔(zero-mode waveguides, ZMWs)和荧光标记在核苷酸焦磷酸链上(Phospholinked nucleotides)。
SMRT Cell含有纳米级的零模波导孔,每个ZMW都能够包含一个DNA聚合酶及一条DNA样品链进行单分子测序,并实时检测插入碱基的荧光信号。ZMW是一个直径只有10~50 nm的孔,当激光打在ZMW底部时,只能照亮很小的区域,DNA聚合酶就被固定在这个区域。只有在这个区域内,碱基携带的荧光基团被激活从而被检测到,大幅地降低了背景荧光干扰。

SMRT Cell和ZMWs
将荧光染料标记在核苷酸的磷酸链而不是碱基上,当核苷酸掺入到新生的链中,标记基团就会自动脱落,减少了DNA合成的空间位阻,维持DNA链连续合成,延长了测序读长。SMRT测序最大限度地保持了聚合酶的活性,是最接近天然状态的聚合酶反应体系。
SMRT测序采用四色荧光标记的dNTP和ZMW孔完成对单个DNA分子的测序:当DNA模版被DNA聚合酶捕获后,4种不同荧光标记的dNTP通过布朗运动随机进入检测区域并与聚合酶结合,与模板匹配的碱基生成化学键的时间远远长于其他碱基停留的时间。因此统计荧光信号存在时间的长短,可区分匹配的碱基与游离碱基。
4色荧光标记4种碱基(即dNTP),在碱基配对阶段,不同碱基的加入,会发出不同光,根据光的波长与峰值可判断进入的碱基类型。每个ZMW孔中,单个DNA分子模板与引物结合,然后与DNA聚合酶结合,被固定到ZMW孔底部。加入四色荧光标记的dNTP后,DNA合成开始。受碱基配对原则支配,连接上的dNTP会在ZMW孔底部停留较长时间,激发后所发出的对应荧光信号被识别,返回的荧光信号会形成一个特殊的脉冲波。另一方面由于荧光信号连接在dNTP的磷酸基团上,当上一个dNTP合成后,磷酸基团自动脱落,保证了检测的连续性,提高了检测速度,并配合高分辨率的光学检测系统以实现实时检测。
CCS测序模式:PacBio测序中,当插入片段(Reads of Insert)< PacBio测序读长(Polymerase Reads)时,测序模式为CCS,插入片段将会被测到N次,PacBio测序为随机错误模式,N次测序结果互相校对,准确率得到极大提升(>99.9%)。如图所示:
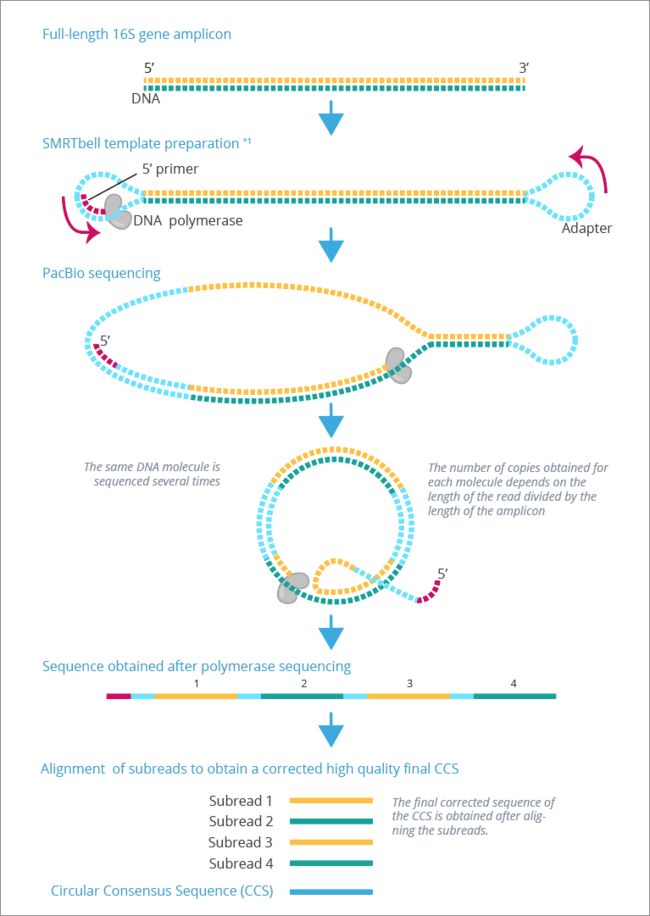
全长16S rRNA基因测序分析流程
质量控制
可以结合Fastp、NGS QC Toolkit、FASTQC等软件进行质量控制。
通常CCS reads质量可以从以下两个方面控制:
(1)测序所得的16S rRNA CCS reads的长度应该为全长或者接近全长,远远低于或者超过预期长度的片段应该过滤;
(2)通常采用CCS reads所有碱基平均质量值( 例如Q20)作为过滤条件;过滤包含模糊碱基(N)的序列。
过滤嵌合体
16S rRNA基因序列扩增和SMRT测序过程中,依然不可避免地会产生嵌合体序列(Chimera),因此需要过滤嵌合体。16S rRNA基因序列测序片段显著增加,嵌合体序列的识别率也将得到提升。UCHIME仍然是嵌合体检测使用最普遍的软件,结合GOLD等数据库可以完成有参的嵌合体检测分析。也可以使用UCHIME基于序列丰度的de novo检测方法识别嵌合体序列。
OTUs聚类与注释
获得预处理的CCS reads之后,按97%相似度进行OTUs聚类分析,可以选择mothur或者QIIME等软件。
对全长16S rRNA基因进行注释时,基于朴素贝叶斯分类器的RDP-Classifier依然是最有效的工具。而在数据库的选择上,可以使用更新最快最全的Silva数据库。
rDnaTools分析软件
rDnaTools是一个分析来自PacBio的16S rRNA数据的工具流程的python包。rDnaTools主要是将Mothur的工具打包,实现了数据导出、过滤和16S序列聚类等。
虽然rDnaTools主要用于分析16S rRNA,但同样适用于18S、23S或其序列,前提是要有合适的参考数据库。
rDnaTools: https://github.com/PacificBiosciences/rDnaTools

文章实例
标题:利用三代测序技术实现“高分辨率”解析菌群组成谱
Singer, E., Bushnell, B., Coleman-Derr, D., Bowman, B., Bowers, R.M., Levy, A., Gies, E.A., Cheng, J.-F., Copeland, A., Klenk, H.-P., et al. (2016). High-resolution phylogenetic microbial community profiling. ISME J 10, 2020-2032.
研究背景:
过去十多年中,随着短读长、高通量测序的兴起,以16S rRNA基因部分可变区序列为目标的分析方法,逐渐替代了基于rRNA基因全长克隆文库的一代Sanger测序法,使我们能对菌群组成进行深度定量解析。然而,由于二代测序读长短的特性,我们无法对测得物种进行精确的分类鉴定,从而限制了我们对菌群代谢功能的深入理解。随着三代SMRT测序技术的出现,我们有望从根本上解决这一瓶颈,实现对rRNA基因全长序列的高通量精准测序。
研究方法:
样本来源:23种细菌和3种古菌组成的人工菌群,以及取自湖水的天然微生物群落样本
测序平台:PacBio RS II(16S rRNA基因全长)+Illumina MiSeq(16S rRNA基因V4区)
对两种测序平台的测序结果进行多样性组成谱分析,并通过计算机模拟,比较16S rRNA基因全长序列和单V区序列的物种分辨能力。
研究结果:
根据两种平台的测序结果,门水平的菌群组成较为相似,但在更精细的水平存在差异。同时,三代测序的结果显著降低了物种分类信息的不确定性。计算机模拟的结果也表明,短读长的单V区测序可能严重低估某些特定类群的微生物,比如水体样本中参与氮循环和甲烷代谢的物种。因此,基于三代测序的菌群多样性组成谱分析能极大地提升物种分类鉴定的精确性,实现“高分辨率”检测的同时,也为深入阐释菌群的代谢功能奠定了基础。

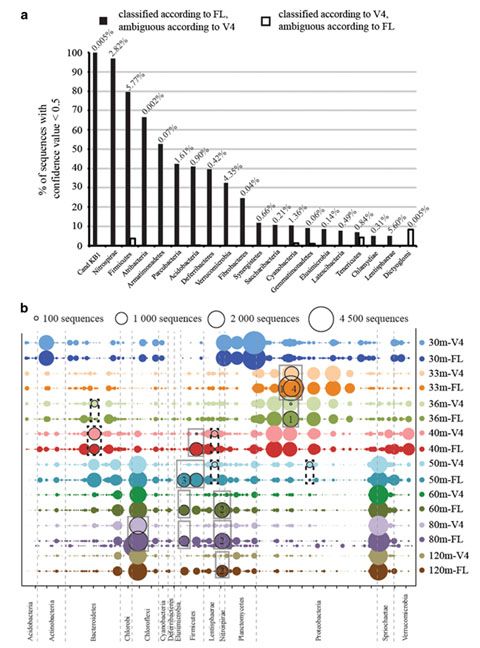
结论:
综上所述,运用三代测序技术进行菌群多样性组成谱解析,将获得更为精准全面的结果。
感谢您的阅读,欢迎点赞、评论、支持和转发!!
