基因组组装完成之后,就需要对最后的质量进行评估。我们希望得到的contig文件中,每个contig都能足够的长,能够有一个完整的基因结构,归纳一下就是3C原则:
- 连续性(Contiguity): 得到的contig要足够的长
- 正确性(Correctness): 组装的contig错误率要低
- 完整性(Completeness):尽可能包含整个原始序列
但是这三条原则其实是相互矛盾的,连续性越高,就意味着要处理更多的模糊节点,会导致整体错误率上升,为了保证完全的正确,那么就会导致contig非常的零碎。此外,这三条原则也比较定性,我们需要更加定量的数值衡量,目前比较常用的标准是N50和BUSCO/CEGMA。
最近有一篇文章"Assessing genome assembly quality using the LTR Assembly Index (LAI) "提出用长末端重复序列来评估基因组完整度,因为LTR比较难以组装,于是就用作评估结果的一个参数了。那问题来了,什么是LTR序列,LTR是在原病毒(整合的反转录病毒)两末的重复序列,结构见下图
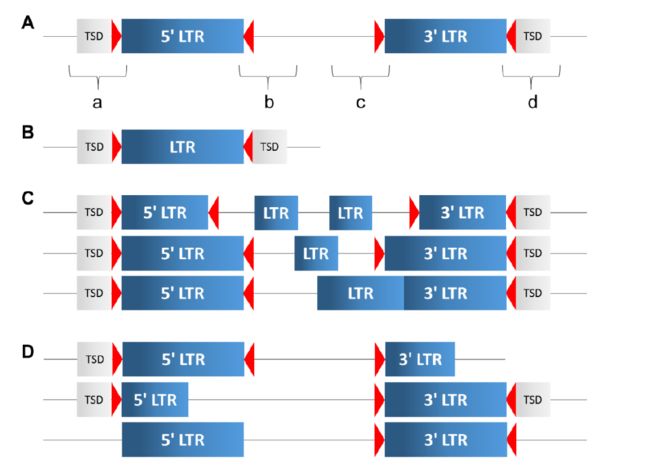
上图中TSD表示target site duplications,红色三角表示LTR motif。A图是一个完整的LTR结构,其中a,b,c是LTR_retriever的分析目标。
LAI指数就是完整LTR反转座子序列占总LTR序列长度的比值。
其实作为一个农学出身,看到LAI,我脑海就想到了Leaf Area Index(叶面积指数)
本文以拟南芥的基因组为例来测试一下这个软件
软件安装
要想保证软件能够顺利的安装,需要先安装如下这几个软件, 好消息是这些软件都可以通过bioconda解决
- makeblastdb, blastn, blastx
- cd-hit-est
- hmmserch
- RepeatMasker
然后从GitHub上下载软件
cd ~/opt/biosoft
git clone https://github.com/oushujun/LTR_retriever.git
进入LTR_retriever文件下修改paths文件,提供每个软件所在的文件路径,下面是我的配置,你需要按照实际所在路径来设置
BLAST+=/home/xuzhougeng/opt/biosoft/ncbi-blast-2.7.1+/bin/
RepeatMasker=/home/xuzhougeng/opt/biosoft/RepeatMasker/
HMMER=/home/xuzhougeng/opt/anaconda2/envs/maker/bin/
CDHIT=/home/xuzhougeng/opt/anaconda2/envs/assembly/bin/
此外,你还需要安装GenomeTools或者LTR_FINDER,或者MGEScan_LTR才能提取出LTR序列,我这里下载的是LTR_FINDER
cd ~/opt/biosoft
git clone https://github.com/xzhub/LTR_Finder.git
cd LTR_Finder/source/
make
软件使用
第一步让我们用LTR_FINDER找到基因组的LTR序列
~/opt/biosoft/LTR_Finder/source/ltr_finder -D 20000 -d 1000 -L 700 -l 100 -p 20 -C -M 0.9 Athaliana.fa >Athaliana.finder.scn
这里的-D表示5'和3'LTR之间的最大距离,-d表示5'和3'LTR之间的最小距离,-L表示5'和3'LTR序列的最大长度,-l表示5'和3'LTR序列的最小长度,-p表示完全匹配配对的最小长度,-C表示检测中心粒(centriole)删除高度重复区域,-M表示最小的LTR相似度。如果不怎么该怎么设置就用默认值。
第二步运行LTR_retriever根据LTR_FINDER的输出识别LTR-RT,生成非冗余LTR-RT文库,可用于基因组注释
~/opt/biosoft/LTR_retriever/LTR_retriever -threads 4 -genome Athaliana.fa -infinder Athaliana.finder.scn
这里的-infinder表示输入来自于LTR_FINDER,它支持同时输入LTRharvest的输出(-inharvest)和 MGEScan-LTR 的输出(-inmgescan). 嫌速度太慢,可以用-threads增加线程数
这一步会调用RepeatMasker,而RepeatMasker要求序列ID长度不大于50个字符,所以请在第一步的时候请先对ID进行修改。
第三步计算LAI。如果前面找到LTR序列太少,低于5%,这一步程序就会报错,那么你就需要调整第一步参数,可能是太严格了。
/opt/biosoft/LTR_retriever/LAI -t 10 -genome Athaliana.fa -intact Athaliana.fa.pass.list -all Athaliana.fa.out
这里最后的结果文件为Athaliana.fa.out.LAI, 第二行就是总体信息,其中RAW_LAI是12.88, LAI是14.47
Chr From To Intact Total raw_LAI LAI
whole_genome 1 119667750 0.0079 0.0612 12.88 14.47
得到的LAI值按照如下评估标准进行分类:
| Category | LAI | Examples |
|---|---|---|
| Draft | 0 ≤ LAI < 10 | Apple (v1.0), Cacao (v1.0) |
| Reference | 10 ≤ LAI < 20 | Arabidopsis (TAIR10), Grape (12X) |
| Gold | 20 ≤ LAI | Rice (MSUv7), Maize (B73 v4) |
和例子一样,TAIR10是中等水平。
参考文献:
- Ou S. and Jiang N. (2018). LTR_retriever: A Highly Accurate and Sensitive Program for Identification of Long Terminal Repeat Retrotransposons. Plant Physiol. 176(2): 1410-1422.
- Ou S., Chen J. and Jiang N. (2018). Assessing genome assembly quality using the LTR Assembly Index (LAI). Nucleic Acids Res. gky730: https://doi.org/10.1093/nar/gky730