笔记内容:
由二代测序产生的序列数据(.fastq)到OTU table, 距离矩阵,物种多样性指数,序列的进化树及物种注释信息的可分析数据,为常规分析流程。可以使用usearch, vsearch, qiime等分析软件实现,在必要的时候需要根据序列信息的具体情况编写脚本予以实现。本笔记描述的是微生物组16S rRNA的数据分析阶段,大概包括哪些内容,如何通过python和R来实现,大概会遇到什么问题以及如何解决。注意本分析是基于OTU table, 样本的距离矩阵,物种多样性指数,序列的进化树及物种注释信息而非基于序列数据(.fastq等)。
包括以下两个内容:input文件介绍
OTU(Operational taxonomic unit),可操作分类单元
各OTU的代表序列(.fasta)
物种注释信息
物种进化树
各样本的多样性指数:α-diversity的input
距离矩阵:β-diversity的input分析内容
物种构成及优势物种
α-diversity
β-diversity
biomarker
功能分析:PICRUSt
input文件介绍
OTU(Operational taxonomic unit),操作分类单元
在二代测序中,每个sample都会测到许多许多序列:
sample1: seq1, seq2, seq3, seq4, seq5...
sample2: seq1, seq2, seq3, seq4, seq5...
sample3: seq1, seq2, seq3, seq4, seq5...
...
每个序列都会有一小段barcode标记,以示它是来自哪个sample。经过一些预先处理,包括去除barcode, 低质量序列,污染序列,嵌合体,等。使用序列聚类算法将相似度(similarity)为97%以上的序列放在一起,组成一个OTU。所以一个OTU内所有的序列均为相似度97%以上的,相似度不足97%的则分到其他的OTU中去。于是我们可以得到OTU_table,一个给出每个sample中每个OTU包含多少reads数目的矩阵:
即每个sample对应每个OTU中的序列reads数目。如sample1在OTU1中有2个序列reads数目。如下所示的OTU_table即丰度。相对丰度则以每个sample(每行)为100%,计算各OTU的reads数目占一个sample中所有的reads数目的百分比。
| sample | OTU1 | OTU2 | OTU3 | OTU4 | OTU5 | ... |
|---|---|---|---|---|---|---|
| sample1 | 2 | 5 | 10 | 13 | 4 | ... |
| sample2 | 2 | 13 | 10 | 13 | 4 | ... |
| sample3 | 2 | 53 | 0 | 1 | 43 | ... |
| ... | ... | ... | ... | ... | ... | ... |
OTU是对相似性序列进行聚类,将海量测序序列聚类成数量较少的分类单元,并且每个OTU提供一个代表序列,基于它进行后续物种注释及分析,更加简便和清晰。
各OTU的代表序列(.fasta)
以下为一个代表序列内容示例:

物种注释信息
将OTU代表序列分别与数据库进行比对,给每个OTU追溯到其物种来源。划分到界(Kingdom)、门(Phylum)、纲(Class)、目(Order)、科(Family)、属(Genus)、种(Species)。虽然不同软件及流程output的物种注释格式可能不一致,但内容大同小异,均包含了上述信息。
你可以结合OTU table和物种注释信息,将相同level的物种丰度相加,整理出每个level的物种丰度文件。比方说将family level中,相同family的物种丰度相加,形成一个family level的物种丰度文件。其作用为可以通过直接比较不同分组的物种丰度,从而找出哪些物种的丰度在组间存在差异,即挑选可以区分不同组的marker.
其形式一般为每个OTU对应以下一条物种注释信息。有些公司测序并初步分析给出OTUtable, 在每行OTU后面直接附上了注释信息。
k__Bacteria; p__Firmicutes; c__Clostridia; o__Clostridiales; f__Lachnospiraceae; g__Robinsoniella; s__peoriensis
物种进化树
为了研究OTU序列所代表的物种进化关系,我们通过OTU代表序列之间的相似性构建物种进化树,代表每个OTU的进化关系。其后缀名为.tree, 可以用figtree软件打开。
各样本的多样性指数:α-diversity的input
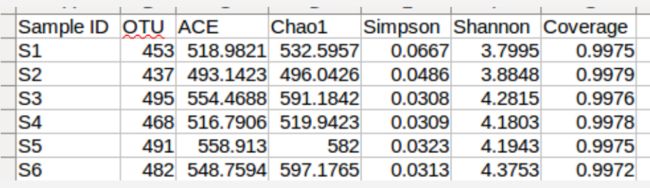
α-diversity是用于回答“一个样本中有多少个物种?”的。所以α多样性指数是针对每个样本的。最简单的一种指数为richness,即每个样本中OTU的个数。
距离矩阵: β-diversity的input

β-diversity是用于回答“两个样本之间的相似程度如何?”这样的问题。它比较两个样本之间的相似度或者差异程度,并给这两个样本计算一个值,通常在0-1之间,以距离矩阵的形式呈现。可以观察到它是对称的,因为两两样本之间相似性的值是一样的。
分析内容
物种构成及优势物种
以下为一篇文章中的示例:
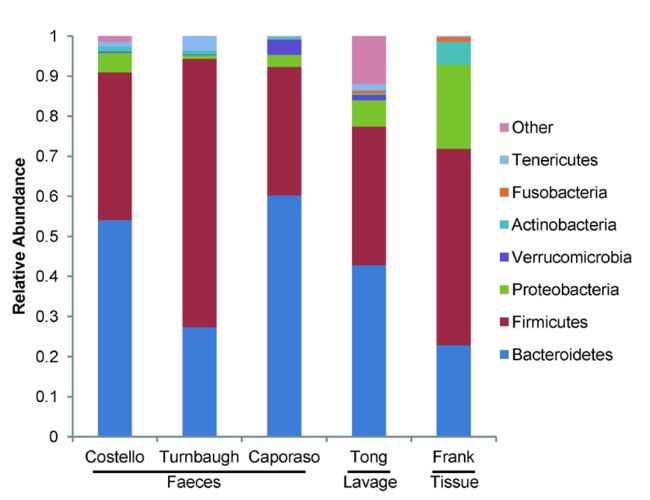
可以大概理解为根据以上5个组的phylum level相对丰度绘制barplot, 观察到相对丰度最大的物种则为优势物种。在上图中为Firmicutes和Bacteroidetes。
α-diversity
包括rarefaction curve, rank abundance curve, 各项多样性指数的组间差异等。
rarefaction curve(稀释曲线)
可参考α diversity分析https://www.jianshu.com/p/7cb452fede5a
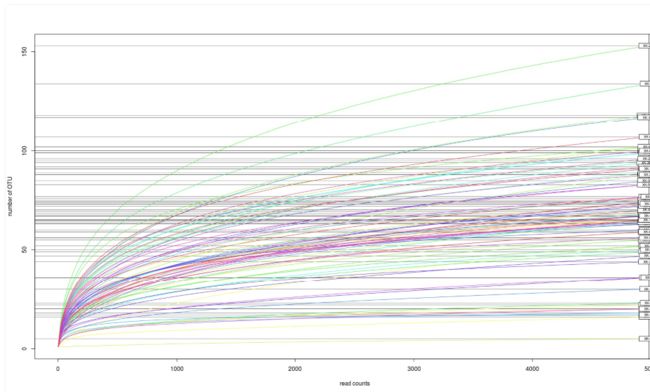
在每个样本中不断抽样,每次都随机抽取一定数量的序列,以抽取到的序列构建OTU。其核心在于resampling。随着抽取的序列数目不断增加,其构建的OTU个数从迅速增加到趋于平坦,则说明抽样的数目合理,更多的序列不会再增加更多信OTU个数。即测序深度达到了要求。 其横轴为每次抽取的read counts, 纵轴为以抽取的read counts构建的OTU个数。 qiime可以生成rarefaction curve, 也可以 用R实现。
rank abundance curve
rank abundance curve比较简单,其横轴为按照相对丰度从大到小排序的OTU的ID(或者其他物种ID),纵轴为相对丰度,如下所示。以下这个例子没有标出X轴的OTU ID, 只注明了其rank(排序)。
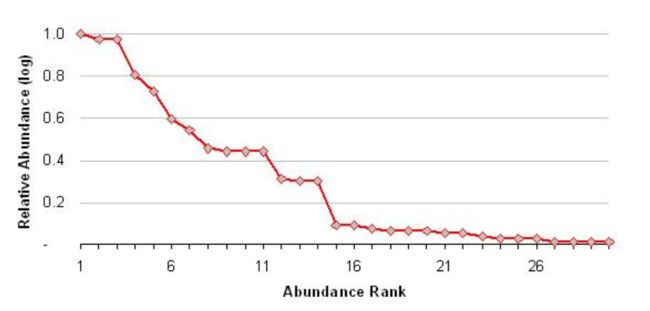
通过它可以了解优势物种有哪些。如果rank abundance curve很陡峭(即一开始很高,然后一个大跳水降很低),说明在样本中有明确的优势物种,且占了很大的比例。拖尾的物种丰度比较稀少。如果它下降的比较平缓,说明各物种都占有一定比例。当然你也可以根据样本分组在一个图中画多个rank abundance curve. 还可以根据一组样本,以相对丰度均值为纵轴画图,如下所示:

各项多样性指数的组间差异
根据各样本的多样性指数:α-diversity的input(即每个样本对应一种多样性指数的数值)和分组信息文件(即根据科学问题,将样本分成不同的类别,而分析的意义在于寻找不同类别之间的差异)。
可以用R将结果可视化。有时候是分多个组,需要注意使用哪种统计方法。可以参考 统计检验简单小结
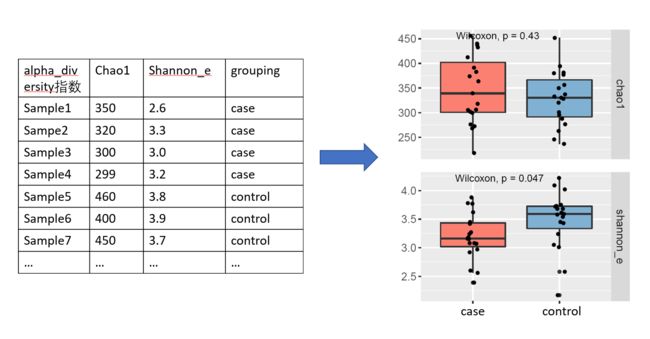
β-diversity
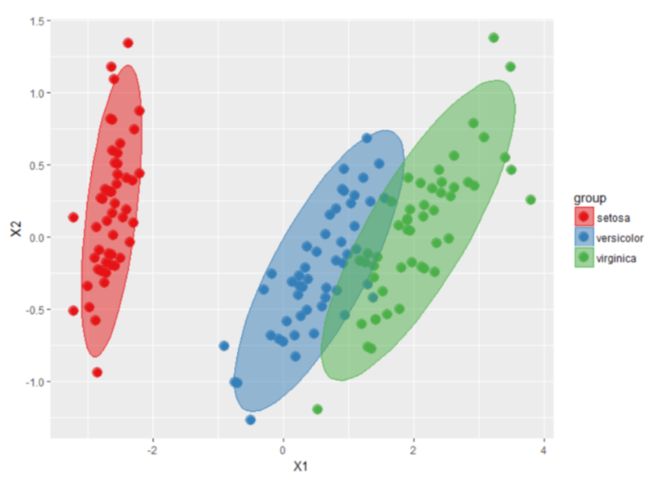
目前常以距离矩阵作为input,使用PERMANOVA做组间差异比较,判断组间物种构成是否有差异,并以主坐标轴分析Principal Coordinates Analysis (PCoA, = Multidimensional scaling, MDS) 产生的新坐标绘图进行可视化。
PCoA(MDS)是根据样本之间的距离,将高维数据投射到低维,达到降维的目的。在PcoA散点图中,聚集在一起的样本相似程度越高,在微生物β-diverisity分析中,即为聚集在一起的样本物种构成更相似。
用R中的ade4及ggplot2绘制PcoA散点图及置信椭圆,具体代码实现见R使用笔记: scatterplot with confidence ellipses;envfit的实现及释意。输出例子如下所示:
PERMANOVA(Permutational (nonparametric) MANOVA)是适用于距离矩阵的非参数统计检验方法, 可以通过R中vegan包的adonis()实现。具体统计原理不做详叙,可以参考这个链接。其在R中使用要点有三个:
- input文件必须为距离矩阵,不是的话需要使用dist()转换。
-
MAN <- adonis(y.std ~ grp, permutations = 1000)实现 - 如果结果p <0.05, 则说明两个组别的物种构成存在差异,且根据R^2值可以知晓这种差异多少比例是由组别的因素贡献的。
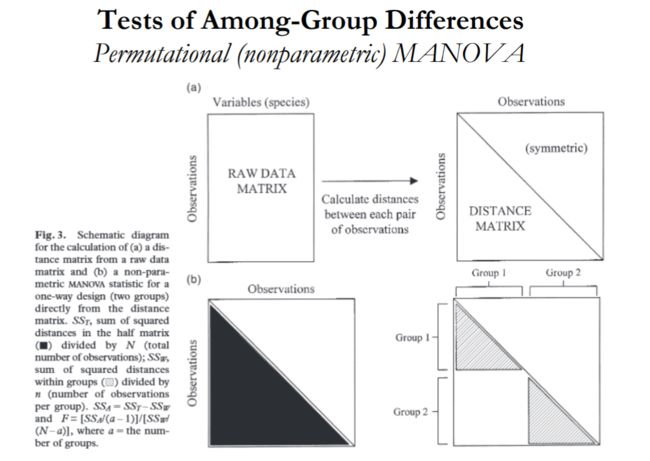

对16s微生物组数据而言,组间物种构成的差异以PERMANOVA的统计检验结果为准,PcoA(MDS)所作的二维或三维散点图为可视化手段,为更直观的展现组间差异。
biomarker
marker, 在微生物组物种层面上,如果某个物种的相对丰度在两组(或多组)间存在有统计学意义的差异,即该物种的丰度高低可以有效区分不同组别,则这个物种为这两组(或多组)的biomarker.
目前我们常用传统统计学方法,boruta及lefse来挑选marker物种。传统的统计学及boruta方法靠python或者R代码完成并可视化,lefse为包装好的软件,可以使用web端版本直接输入input文件,选择参数运行即可得结果,也可以在linux系统中安装lefse软件,在终端运行并得到结果。
传统统计学方法
传统统计学方法则简单的以物种丰度表为Input,按照不同分组,对每个物种的组间进行统计检验,如果组间差异有统计学意义,则该物种为biomarker。以下为一个input及结果展示示例,可以用heatmap及boxplot来了解不同组间物种丰度的差异方向,并使用统计检验(本例中使用t.test)了解差异是否具有统计学意义,并需要做多重统计检验矫正(p.adjust)。用R实现的代码参考如下:
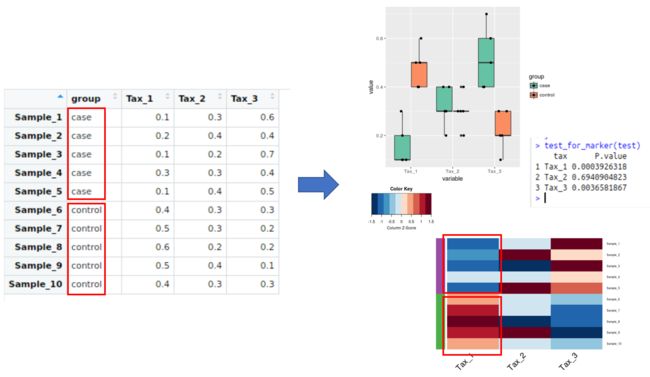
test <- read.csv('test_marker.csv', header = TRUE, row.names = 1, check.names = FALSE)
### heatmap
library(gplots)
library(RColorBrewer)
library(ggplot2)
tax = c('Tax_1','Tax_2','Tax_3')
test_data <- test[,tax]
grouping <- as.factor(test$group)
plot_color <- c(brewer.pal(5, "Set1")[4],brewer.pal(5, "Set1")[3])[grouping]
heatmap.2(as.matrix(test_data),
trace="none",
scale="column",
RowSideColors = plot_color,
dendrogram = "none",
symbreaks = TRUE,
col=rev(brewer.pal(10, "RdBu")), breaks = seq(-1.5,1.5,0.3),
density.info=c("none"),
margins=c(8,16), cexRow = 0.8, srtCol = 45, Colv=FALSE, Rowv = FALSE
)
### boxplot and test for comparint between groups
test_m <- read.csv('test_marker.csv', header = TRUE, check.names = FALSE)
colnames(test_m)[1] = 'ID'
test_m <- melt(test,id.vars = 'group')
ggplot(test_m, aes(x = variable, y = value, fill = group)) +
geom_boxplot(position = position_dodge(0.8)) +
geom_point(position = position_jitterdodge()) +
scale_fill_brewer(palette = "Set2")
test_for_marker <- function(tax_ra){
P.value = c()
tax = c()
tt = colnames(tax_ra)[2:length(colnames(tax_ra))]
for (i in tt){
t <- t.test(tax_ra[[i]] ~ group, data = tax_ra)
P.value = c(P.value, t$p.value)
tax = c(tax, i)
}
p <- data.frame(tax, P.value)
return(p)
}
test_for_marker(test)
当然不一定非要用heatmap和boxplot来展示结果,可视化方法是灵活的,需要根据具体科学问题来变通。
传统统计学方法可能会稍微有些麻烦,但是可以很细致的把每层物种都筛查一遍寻找marker;相比包装好的软件,自主调节一些分析和作图的细节也更加灵活。
boruta和lefse
可以参考boruta:用于特征物种的挑选, 它大概是将每个feature(物种)的数值打乱随机排列,然后看该feature(物种)的原始评分是否比打乱的随机数值更加有规律(显著)。它的好处在于不必拘泥于传统统计学检验方法的先验条件,对于变量特别多,容易过拟合的数据也很友好。
Lefse有在线使用的网站,也可以下载软件到linux系统中在终端使用。它的原理是先使用非参数检验Kruskal-Wallis test检测所有物种在指定组别中的显著性,然后用wilcoxon rank-sum test两两比较。LEfSe采用线性判别分析(LDA)来估算每个物种的丰度对差异效果影响的大小,应该是线性模型的原理。其网页版基本上跟着以上链接上传文件,按照提示指引输入分组信息及各项参数,就可以得到biomarker分析结果。其优点在于方便省心,图好看且使用广泛。
对与marker的挑选没有一个固定的方法标准,比方说什么情况下适合用哪种方法之类的。就使用了16s数据的各种文献来看,用哪种方法的都有。
功能分析:PICRUSt
PICRUST也是可以安装在linux系统上的软件,用于物种功能预测分析。在16s数据中并不十分推荐,因为其在16s中的功能预测有一定局限性。但是目前也有很多文献将功能分析纳入了分析结果中。功能分析仅供参考,需要结合科学问题慎重考虑。
另外也有人做出了分析网站http://www.microbiomeanalyst.ca/faces/home.xhtml可以直接上传符合格式要求的OTU table, meta文件及物种信息就可以输出一系列上文提到的各种分析结果,操作轻松,不用写代码。但是在数据形式不寻常,比方说对于时间序列,单个样本的重复测量结果,分组复杂的数据,需要进行数据预处理的数据(比如需要取对数等)并不能完全依赖包办型的分析网站。不论是代码、软件还是包办的分析网站都只是分析工具,需要根据具体科学问题,合理使用它们。
Ref:
R作图,PERMANOVA的adonis()实现,rank abundance等参考
PcoA(mds)的R实现
生态学数据的多元分析:包括PERMANOVA的学习网站,含原理。