转录组学习一(软件安装)
转录组学习二(数据下载)
转录组学习三(数据质控)
转录组学习四(参考基因组及gtf注释探究)
转录组学习五(reads的比对与samtools排序)
转录组学习六(reads计数与标准化)
转录组学习七(差异基因分析)
转录组学习八(功能富集分析)
对原始测序fq文件数据进行质量控制
任务
了解fastq测序数据
需要用安装好的sratoolkit把sra文件转换为fastq格式的测序文件,并且用fastqc软件测试测序文件的质量!
作业,理解测序reads,GC含量,质量值,接头,index,fastqc的全部报告,
SRA文件解压为fastq原始测序文件
首先就是要将前一步从NCBI的SRA数据库下载的SRA格式的压缩文件 解压缩为原始的测序出来的fq文件。用到的软件就是之前下载的Sratoolkit软件里的fastq-dump程序。
-
fastq.dump.2软件使用参数帮助具体可用命令fastq-dump.2查看,其中常用参数:
- --split-3:现在大多数测序都为双末端测序,此为重要参数。生成结果为*_1.fastq和*_2.fastq
- --gzip: 原始fastq文件直接解压文件太大!1.2G文件能解压出10G的文件!,-gzip为常用的一种压缩方式,生成文件结尾*.fastq.gz,后续也可以直接查看。
- -O:解压出的文件输出路径。
- -A:输入的sra文件。
所以,对于下载的SRA文件从56~62进行解压可以写个简单的循环脚本再加上fastq-dump命令:fastq.dump.2 --gzip --split-3 -O (path) -A (SRA)即可:
fastq-dump
for i in `seq 56 62`
do
fastq-dump.2 --gzip --split-3 -O ./1_raw_data -A SRR35899${i} ##如果出错一定注意输出路径的问题,可以改为文件夹的绝对地址,对于fastq-dump软件也同样适用
done
- 生成文件:
 image
image
fastqc
- 基本参数: -o path(输出路径) --nogroup 不生成单独的文件夹。
利用xargs批量生成fastqc程序脚本
ls *.fastq.gz 会列出所有要操作的文件;xargs 命令通过管道,将上一个命令的输出作为自己的输入;-i 参数的意思是按行处理,并且会将每行的内容存到一个特殊变量 {} 里;echo fastqc -o ./ {} 是要对每一行执行的命令;{} 代表每一行的内容。
- 这么多样品,明早要结果,要并行计算才行 在命令结尾添加 &,注意需要转义,前面加\,并且要nohup &并行处理。注意 echo 是可选的
mkdir 2_fastqc && cd 2_fastqc
ls /sas/supercloud-kong/wangtianpeng/data/biotrainee/1_raw_data/*.fastq.gz | xargs -i echo nohup /usr/local/bin/fastqc -o /sas/supercloud-kong/wangtianpeng/data/biotrainee/2_fastqc/ --nogroup {} \& > fastqc.sh
bash fastqc.sh
注意,此时可能会有 too many levels是因为fastqc软件的软连接的问题,将软连接改为硬地址即可。
- 写法二:简单的循环for脚本也可以
for i in /sas/super_cloud/wangtianpeng/data/1_raw_data/*.fastq.gz
do
nohup fastqc -o ./ --nogroup ${i} \&
done
- 生成文件结果:
 image
image
fastqc质控结果解读
首先用winscp或者Xftp软件将2_fastqc文件夹下的html文件下载到本地电脑里,可以直接用浏览器打开。
- fasqc质控的结果解读是个很大的坑,软磨硬磨看了不少参考文档才模糊的明白一些指标。
主要参考文章> NGS基础 - FASTQ格式解释和质量评估
FastQC 你需要知道的在这里!
数据质量不好,可以用么?
我的基因组(十):测序数据质量控制
转录组之数据质控
PANDA姐的转录组入门(3):了解fastq测序数据
静渊的学习日志
-
Fastqc报告整体浏览
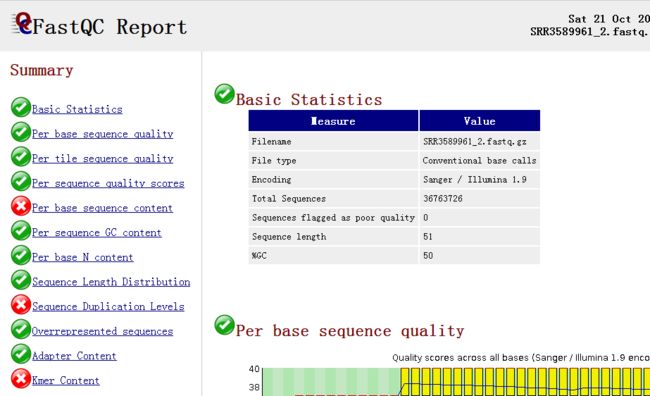 image
image- SUMMARY: 绿色合格,黄色警戒,红色不合格。总体来说,并不是都是绿色就代表合格,红色就代表放弃数据,还需要查看后面具体的项目内容。此报告结果有三项不合格,需要接着看。
- Basic Statistics: 测序平台illumina,总的reads数目(和覆盖度有关)36M左右,reads长度51bp,GC百分比 50%。基本符合。
-
某一位置上所有读段的测序质量评分Per Base Sequence Quality
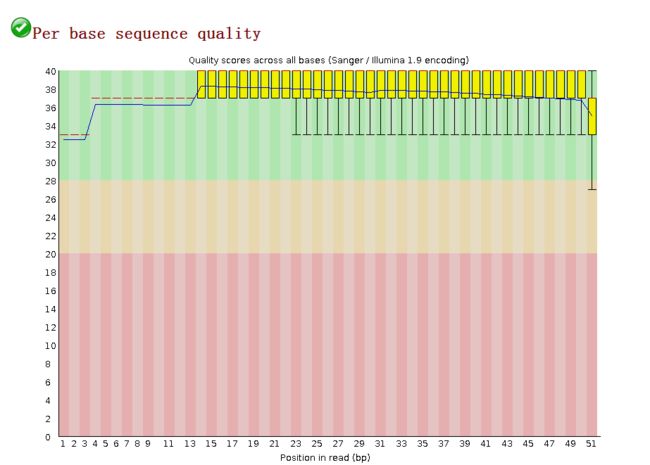 image
image- quality就是Fred值,一条reads某个位置上出错概率为0.01时,quality值就是20,即常说的Q20。
- 就是一个箱线图boxplot,黄色箱子(25%和75%的分数线),红色线(中位数),蓝线是平均数,下面和上面的触须分别表示 10 % 和 90 % 的点
- 横坐标reads的碱基位置,最大值即为读长,纵坐标代表质量的好坏(判断的准确性)
- 如果任何一个位置的下四分位数小于10或者中位数小于25,会显示“警告”;如果任何一个位置的下四分位数小于5或者中位数小于20,会显示“不合格”
- 根据此次数据结果所看,基本处于绿色合格位置,测序质量不错!
-
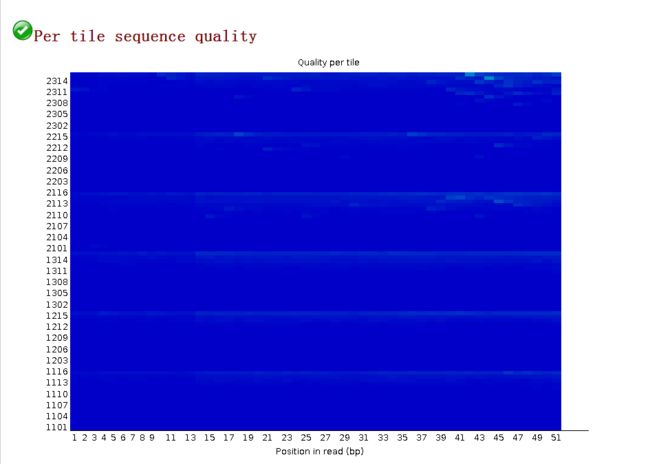
每次荧光扫描的质量Per Tile Sequence Quality
 image
image- 图中横轴代表碱基位置,纵轴代表 tile 编号.
- 图中的颜色是从冷色调到暖色调的渐变,冷色调表示这个 tile 在这个位置上的质量值高于所有 tile 在这个位置上的平均质量值,暖色调表示这个 tile 的在这个位置上的质量值比其它 tiles 要差;一个很好的结果,整张图都应该是蓝色,简单来说,就是看图内有无除蓝色外的亮点,有亮点代表低于平均值
- 根据此结果看,基本合格。
-
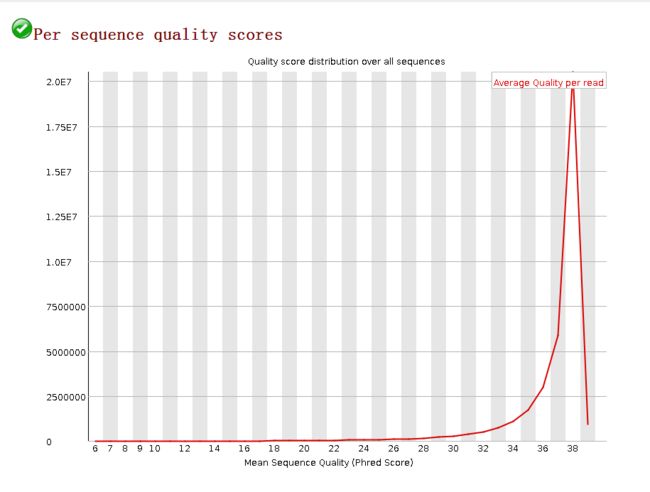
每条序列平均碱基质量的分布Per Sequence Quality Scores
 image
image- 图中横轴为测序质量值,纵轴为 reads 数量;
- 看 横坐标值分布、红色曲线走势,峰的位置。红线上的每一个点表示quality值所对应的reads的数量,其面积就是总的reads数。
- 如果最高峰所对应的横坐标质量值小于 27 (错误率 0.2 %) 则会显示“警告”,如果最高峰的质量值小于 20 (错误率 1 %) 则会显示“不合格”。
- 如图所示红线单峰,分值在37左右,所以reads很可靠。
-
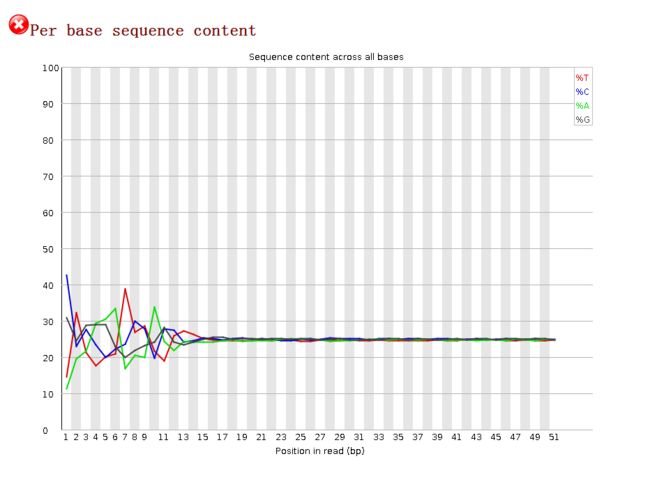
每个位置的4种碱基组成比例Per Base Sequence Content
 image
image- 这项看的是每个位置上ATCGG的比例。一个完全随机的文库内每个位置上 4 种碱基的比例应该大致相同,因此图中的四条线应该相互平行且接近25的位置左右;
- 在 reads 开头出现碱基组成偏离往往是我们的建库操作造成的,在reads上加接头的碱基组成不是均一的。会造成明显的碱基组成偏离。
- 如果任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 10 % 以上则报“警告”,任何一个位置上的 A 和 T 之间或者 G 和 C 之间的比例相差 20 % 以上则报“不合格”。
- 此结果总体上处于25%说明质量可以,但在前13个bp位置上严重分离,说明有碱基偏向性。可能有接头的污染。
-
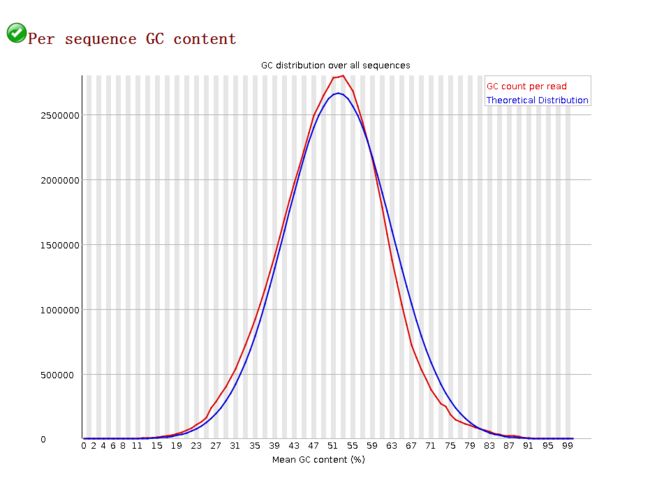
GC含量的分布图:Per Sequence GC Content
 image
image- 在一个正常的随机文库中,GC 含量的分布应接近正态分布,且中心的峰值和所测基因组的 GC 含量一致。由于软件并不知道所测物种真实的 GC 含量,图中的理论分布是基于所测数据计算得来的;
- 如果出现不正常的尖峰分布,则说明文库可能有污染, (如果是接头的污染,那么在 overrepresented sequences 那部分结果还会得到提示),或者存在其它形式的偏选;
- 总体上就是看红色的线与蓝色线正态分布趋势是否接近。此图可知道红色线与蓝色线较为接近,质量较好。
-
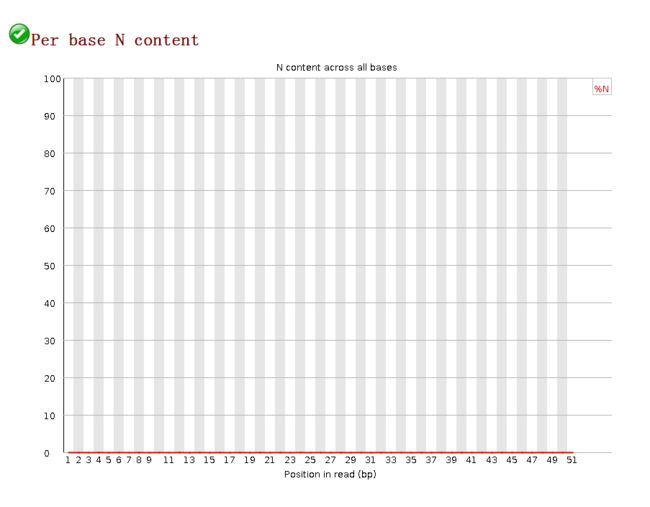
每个位置上N的比例Per Base N Content
 image
image- 在测序仪工作过程中,如果不能正常完成某个碱基的 calling,将会以 N 来表示这个位置的碱基,而不是 A、T、C、G;有时在序列中会出现较低比例的 Ns,尤其是靠近序列末端的位置,这说明系统不能正常的 call 这部分碱基;
- 横坐标表示reads的位置,纵坐标表示N的比例。如果任何一个位置 N 的比例大于 5 % 则报“警告”,大于 20 % 则报“失败”。
- 此图可知基本无N,皆已测得为ATGC的碱基。测序质量较好。
-
reads的长度分布Sequence Length Distribution Content
 image
image- 测序仪出来的原始 reads 通常是均一长度的,但经过质控软件等处理过的数据则不然;经过质控软件处理过的reads长度则不一样。
- 当 reads 长度不一致时报“警告”,当有长度为 0 的 reads 时则报“不合格”。
- 此图可知为测序仪产出的reads,长度皆为51bp
-
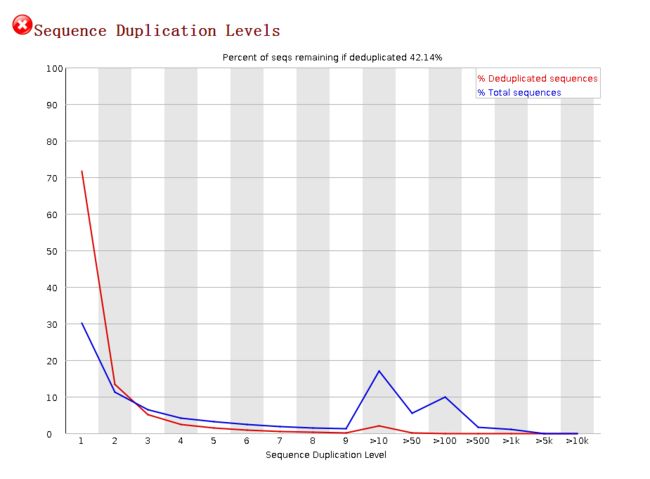
序列重复的水平Sequence Duplication Levels
 image
image- 图中横轴代表 reads 的重复次数 ( 1 表示 unique 的序列,2 表示有 2 条完全相同的 reads ...),大于 10 次重复后则按不同的重复次数合并显示。 纵坐标表示各重复次数下的 reads 数占总 reads 的百分比;
- 蓝线展示所有 reads 的重复情况,红线表示在去掉重复以后,原重复水平下的 reads 占去重后 reads 总数的百分比;如果非 unique 的 reads 占总 reads 数的 20 % 以上则报 ”警告“,占总 read 数的 50 % 以上则报 ”不合格“。
- 看了不同的文档说明皆表示:不合格报错对于此项是正常现象,不需要太过关注。
-
大量重复出现的序列Overrepresented Sequences
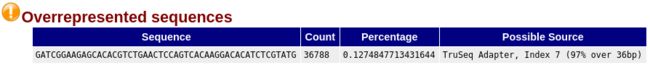
- 显示同一条 read 出现次数超过总测序 reads 数的 0.1 % 的统计情况。正常文库内序列的多样性水平很高,不会有同一条 read 大量出现的情况,这部分结果会把大量出现的 reads 列出来,并给出可能来源;
- 如果有任何 read 出现的比例超过总 reads 数的 0.1 % 则报 '警告',超过总 reads 数的 1 % 则报 '不合格'。
- 如果检测出一条多重复序列,重复次数较多,推测可能是TrueSeq接头序列。
-
每一位置上是接头序列的比例Adapter Content

- 显示 reads 中的接头含量,并显示可能的来源。图中横轴为碱基位置,纵轴为含有接头序列的比例;
- 正常的情况下接头的含量应该接近 0,如果 reads 中的接头含量过高,说明文库内小片段比例偏高 (这个可以从文库质检报告中看出来),这可能是由于片段选择时选取的长度偏短或者使用切胶的方式回收片段时上样过多致使小片段不能很好的分离等原因造成的;如果接头的含量随着碱基的位置增大而逐渐升高,则表示 reads 中含有接头 (如图所示),这部分接头会影响后续的分析,我们需要截掉 reads 中的接头序列或者将含有接头的 reads 完全删除。
- 如果任何重复 read 超过总 reads 数的 5 % 则报 '警告', 超过总 reads 数的 10 % 则报 '不合格,
- 由图可知测序是没有接头污染的。如果有接头污染,在序列尾端会出现一个上扬的曲线。
-
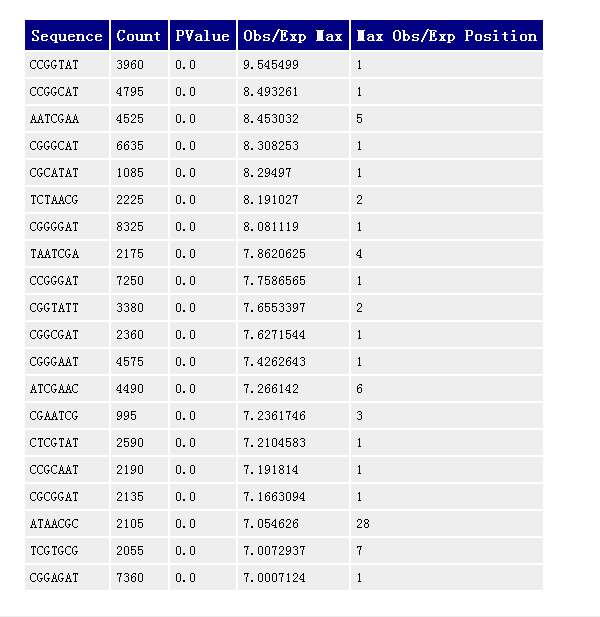
Kmer Content
 image
image
 image
image- 这部分分析假定任何特定长度的短序列在正常的文库中不会有明显的位置偏好性,即使有特殊的生物学原因影响到 Kmers 分布,那在同一序列不同位置上的概率也应该是相同的;
- 如果任何 Kmer 的 P 值小于 0.01 则 '警告',小于 0.00001 则 '不合格'。
- 短序列的重复性,这部分还是不是很明白啥意思,看不下去了,以后如果遇到不合格情况多的时候再看吧,感觉不是个很重要的指标??
他人经验介绍
碱基质量值随着 reads 的位置的增大而降低是正常现象,且通常反向 reads 的质量要比正向的差。
Hiseq 2000 以后的测序仪,前10 - 15个循环的碱基质量要比 reads 中部的碱基质量值低。
对于所有的 Illumina 平台,碱基质量的中值应该在 30 以上,如果同一位置碱基质量值的变异很大,表明文库中存在低质量的样本。
序列碱基质量值变低,往往也会同时表现出碱基组成的异常。
如果碱基质量值有一个骤然的降低,通常表明有接头的污染。
GC 含量有很高的偏离,同样也暗示可能存在一定的污染。
这里所说的污染,可以根据各部分结果综合判断是什么引起的污染,通常表现的是测序接头的污染。
样本的种类和其本身质量、建库方式、测序平台等很多方面都会影响到最终的测序质量。质量检测只是用来查看一下文库的质量,让我们对数据有一个基本的认识,并不是看起来质量不好的文库就一定不能用来做进一步的分析。个人觉得只要确定数据是来自目标样本,没有其它材料的污染,则可通过进一步的质量控制去接头、去低质量的 reads 等来控制数据质量,进行接下来的分析。然后综合其它分析结果来判断样本到底是否可用。
利用multiqc对fastqc结果综合解读
作用:由于fastqc结果会生成多个qc文件报告,一个个点开太麻烦了。所以本着能偷懒就绝不重复劳作的原则,找到了此multiqc软件把多个测序结果的fastqc结果整合成一个报告。
安装,利用conda
conda install -c bioconda multiqc
multiqc
- 具体使用
1. 先进入到之前用fastqc得到测序的html文件和*fastqc.zip文件夹列表里。
2. 直接简单粗暴的命令
multiqc *fastqc.zip --pdf
- 结果会生成一个multiqc文件夹和一个html文件,传输到本地电脑上浏览器打开即可:
结果如图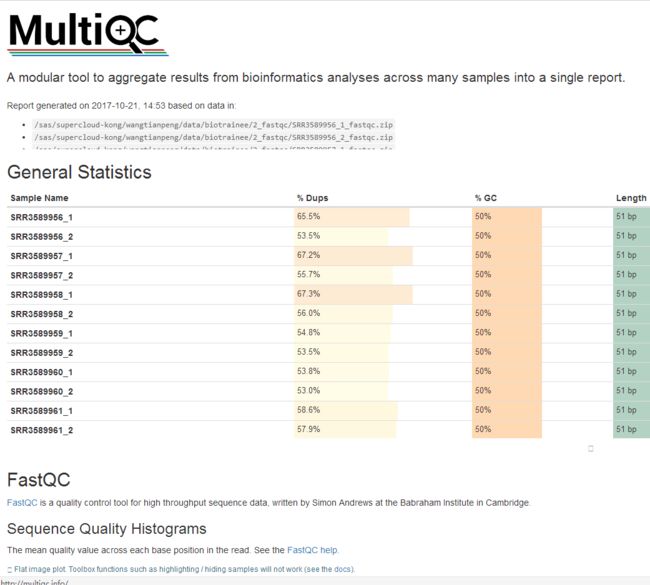 image
image
一目了然,多棒!!
自己写Perl脚本统计Q20,Q30等指标
- fastqc和multiqc的结果是用图形化的方式展示测序的质量,但任然没有给具体q20,q30指标。此时就可以自己根据fastqc结果文件里的数据来自己写个脚本来统计Q20,Q30,GC含量,reads数目。
- 整体思路:
- 首先是perl里的文件操作,涉及DIR,readdir,循环操作符next unless, 循环跳出last if ,perl里调用系统shell命令grep获取GC%和总的reads数, Q20及Q30的计算(1-(前20之和/总和))
- 参考并仿写perl脚本:
#!/usr/bin/perl -w
opendir (DIR, "./") or die "can't open the directory!";
@dir = readdir DIR;
foreach $file ( sort @dir) {
next unless -d $file;
next if $file eq '.';
next if $file eq '..';
$total_reads= `grep '^Total' ./$file/fastqc_data.txt`;
$total_reads=(split(/\s+/,$total_reads))[2];
$GC= `grep '%GC' ./$file/fastqc_data.txt`;
$GC=(split(/\s+/,$GC))[1];
chomp $GC;
open FH , "<./$file/fastqc_data.txt";
while (){
next unless /#Quality/;
while (){
@F=split;
$hash{$F[0]}=$F[1];
last if />>END_MODULE/;
}
}
delete $hash{">>END_MODULE"};
$all=0;$Q20=0;$Q30=0;
$all+=$hash{$_} foreach keys %hash;
$Q20+=$hash{$_} foreach 0..20;
$Q30+=$hash{$_} foreach 0..30;
$Q20=1-$Q20/$all;
$Q30=1-$Q30/$all;
print "$file\t$total_reads\t$GC\t$Q20\t$Q30\n";
}
- 结果:
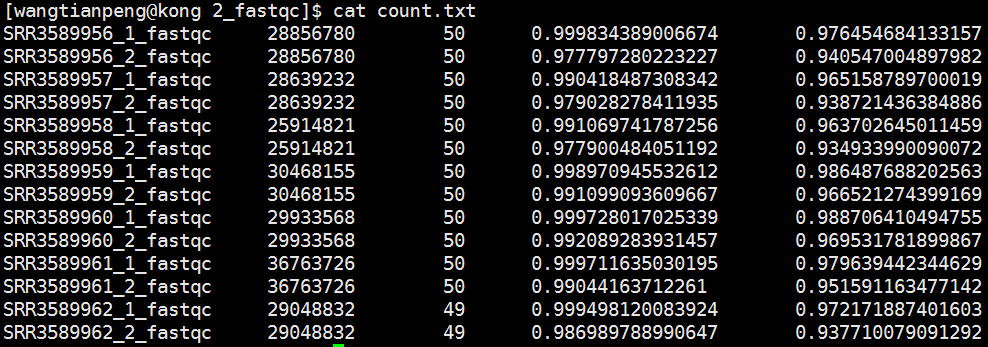 image
image
可以看出Q20指标基本在98/99左右,而Q30指标在95左右,可以说质量非常好了! 另外根据他人的一个经验指标:
如果说Q20的reads可以达到95%以上,Q30的reads达到85%以上,我们认为本次测序还可以。当然我们可以通过过滤提高高质量reads的比率,测序量需达到要求。
2017/10/22下午3点小结:通过此次学习,对fastqc质量报告各项指标的意义有了较为深刻的理解,另外对一些循环脚本,xargs的使用以及一个统计其q20、q30的的perl脚本进行了学习并仿写。目前还只是前期的数据基础整理,还未到转录组分析核心部分,慢慢来吧。
参考文章
- NGS基础 - FASTQ格式解释和质量评估(https://mp.weixin.qq.com/s?__biz=MzI5MTcwNjA4NQ==&mid=2247484047&idx=1&sn=3e2a79d9f56040a57ac2e16cf1923b54&chksm=ec0dc705db7a4e133e6e91de9ac11a6c0690f03d7b3b760b358e9b0d1a2d57974599419d9fff&scene=21#wechat_redirect)
- FastQC 你需要知道的在这里!(https://mp.weixin.qq.com/s?__biz=MzI4NjMxOTA3OA==&mid=2247484306&idx=1&sn=2d1d0734578209379d69e6cdfa133edc&chksm=ebdf8b1bdca8020d7d4d615b16ba06b96d725d5104ffbf8ed8fe9490b77c639381a7c8d0c3b9&scene=21#wechat_redirect)
- 数据质量不好,可以用么?(https://mp.weixin.qq.com/s?__biz=MzI4NjMxOTA3OA==&mid=2247484343&idx=1&sn=a385feaed19be0d2180ca017a0079fbf&chksm=ebdf8b3edca80228b97cef24cb91d3d8ff10f2ac1ac3e69a91e571f7fd48e4756c952fa640b9&mpshare=1&scene=1&srcid=0713hgFPt6wMyWCuxuJyx0Pd#rd)
- 我的基因组(十):测序数据质量控制(https://mp.weixin.qq.com/s/q73DquRCEj7saiiccUL9qw)
- 转录组之数据质控(http://www.jianshu.com/p/2ed3622ed4a8)
- PANDA姐的转录组入门(3):了解fastq测序数据(http://www.biotrainee.com/thread-1853-1-1.html)
- 静渊的学习日志(http://yanshouyu.blog.163.com/blog/static/214283182201302835744453/)