qRT-PCR实验原理
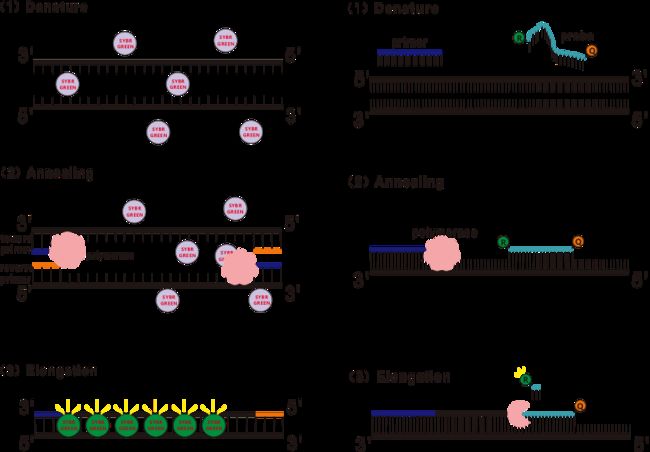
RT-qPCR由普通PCR技术发展而来,它是在传统PCR反应体系中加入荧光化学物质(荧光染料或者荧光探针),根据各自不同的发光机制实时检测PCR退火、延伸过程中荧光信号变化来计算PCR每个循环中产物的变化量,目前最常见的方法为荧光染料法和探针法。
荧光染料法:
一些荧光染料如SYBR Green Ⅰ,PicoGreen,BEBO等,它们本身不发光,但结合于dsDNA的小沟后会发出荧光。所以当PCR反应刚开始时机器并不能检测到荧光信号,当反应进行到退火-延伸(二步法)或者延伸阶段(三步法),此时双链打开,在DNA聚合酶的作用下新链合成,荧光分子就结合于dsDNA的小沟中并发出荧光,随着PCR循环数的增加越来越多的染料与dsDNA结合,荧光信号也不断的增强。以SYBR Green Ⅰ为例。
探针法:
Taqman探针是最为常用的一种水解探针,在探针的5’端存在一个荧光基团,通常为FAM,探针本身则为一段与目的基因互补的序列,在探针的3’端有一个荧光猝灭基团,根据荧光共振能量转移原理(Förster resonance energy transfer, FRET),当报告荧光基团(供体荧光分子)和猝灭荧光基团(受体荧光分子)激发光谱重叠且距离很近时(7-10nm),供体分子的激发可以诱发受体分子发荧光,而自身荧光减弱。所以PCR反应开始,探针游离于体系中完整存在时,报告荧光基团并不会发出荧光,当退火时,引物和探针结合于模板,在延伸阶段,聚合酶不断的合成新链,由于DNA聚合酶具有5’-3’核酸外切酶活性,到达探针时,DNA聚合酶就会将探针从模板上水解下来,报告荧光基团和猝灭荧光基团分开,释放荧光信号。由于探针和模板存在一对一的关系,所以在试验的精度和灵敏度上,探针法都要优于染料法。
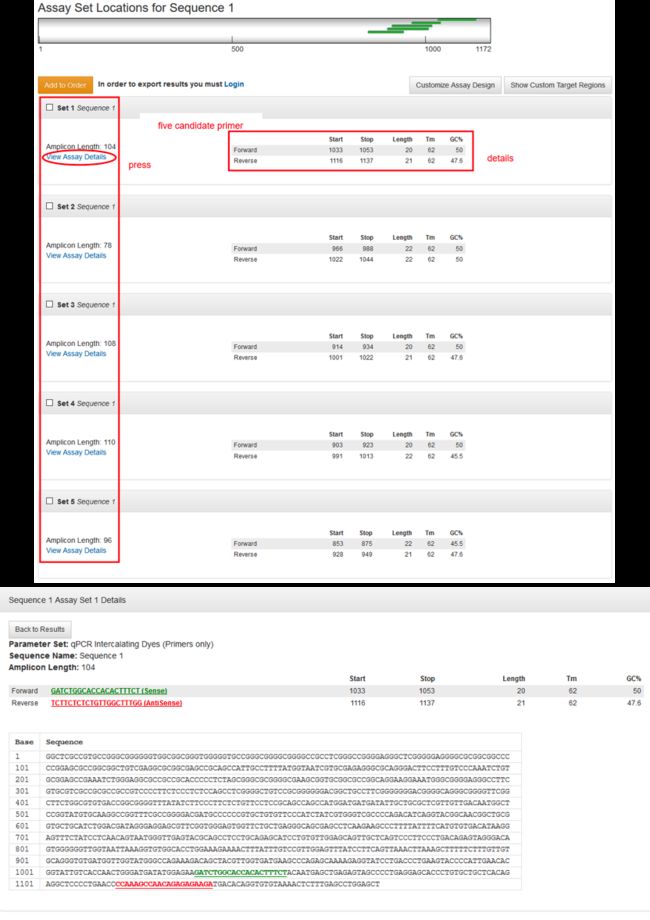
引物设计
原则:
引物应用核酸系列保守区内设计并具有特异性。最好用cDNA序列,mRNA序列也可,如果没有则找出DNA序列的cds区域设计。
做荧光定量产物长度80-150bp最好,最长是300bp
引物长度一般在17-25碱基之间,上下游引物不能相差太大。
G+C含量在40%~60%之间,45-55%最佳。
TM值在58-62度之间。
尽量避免引物二聚体和自二聚体,(不要出现超过4对连续互补的碱基)发卡结构,如果不可避免,使ΔG<4.5kJ/mol*
如果不能确保反转录过程中gDNA已经除干净,最好设计出夸内含子的引物*
3′端不可修饰,而且要避开AT,GC rich的区域,避开T/C,A/G连续结构(2-3个)
引物与非特异性扩增序列的同源性最好小于70%或者有8个互补碱基同源。
数据库:
CottonFGD search by keywords
引物设计:
IDT-qPCR primer design

ncRNA引物设计:
lncRNA: 和mRNA的步骤一致。
miRNA:参考这个方法:茎环法miRNA引物设计-丁香园 茎环法原理:由于miRNA全部为23nt左右的短序列,无法进行直接PCR检测,所以使用了茎环序列这个工具,茎环序列是一段50nt左右单链DNA,可以自身形成发卡结构,3’末端可以设计成与miRNA部分片段互补的序列,那么在反转录时就可以将目的miRNA连接到茎环序列上,那么总长度就达到70bp,就符合qPCR确定的扩增产物的长度。加尾法miRNA引物设计。
扩增特异性检测:
Online blast database:CottonFGD blast by sequence similarity
Local blast:参照我之前写的用Blast+做本地blast,linux和macos可以直接建立本地数据库,win10系统需要安装ubuntu bash后也可以做。本地blast数据库建立及本地blast;win10开启ubuntu bash。
Notice:陆地棉海岛棉为四倍体作物,所以blast结果往往会是两个或多个匹配,以往经验以NAU的cds作为database进行blast很可能找到两个只有几个SNP差异的同源基因,通常通过引物设计是分不开这两个同源基因的,所以就将他们当做同一个来做,如果有明显的indel,通常把引物设计在indel上,但这样就可能导致引物的二级结构的自由能变高,导致扩增效率的下降,但这是不可避免的。
引物二级结构的检测:
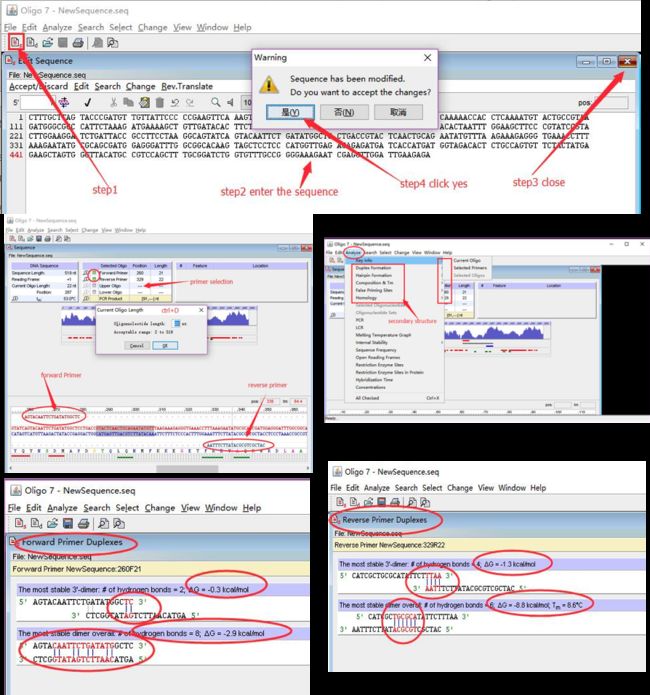
步骤: 打开oligo 7→输入模版序列→关闭子窗口→保存→将引物定位到模版上,按ctrl+D设定引物长度→分析各种二级结构,例如自二聚体,异二聚体,发卡,错配等。图4中最后两张图片为引物测试结果,前引物结果较好,没有明显的二聚体和发卡结构,没有连续的互补碱基,且自由能绝对值也小于4.5,而后引物则出现了连续6碱基互补,自由能为8.8;另外在3’端也出现了较为严重的二聚体话,出现了连续4碱基的二聚体,虽然自由能不高,但3’的二聚体化可以严重的影响扩增特异性和扩增效率。此外还需要对发卡及异二聚体,错配进行检查。
扩增效率检测:
PCR反应的扩增效率严重的影响到PCR的结果,同样在qRT-PCR中,扩增效率对定量的结果尤为重要,除去反应液(reaction buffer)中其他物质及机器和protocol带来的影响,引物的质量对于qRT-PCR的扩增效率影响也非常大,为了保证结果的准确性,无论是相对荧光定量还是绝对荧光定量均需要对引物的扩增效率进行检测,而公认的有效的qRT-PCR的扩增效率为85%-115%之间有两种方法:
1.标准曲线法:
a.将cDNA混合
b.梯度稀释
c.qPCR
d.线性回归方程计算扩增效率
2.LinRegPCR
LinRegPCR is a program for the analysis of real time RT-PCR Data,also called quantitative PCR(qPCR)data based on SYBR Green or similar chemistry. The program uses non-baseline corrected data,performs a baseline correction on each sample separately,determines a window-of-linearity and then uses linear regression analysis to fit a straight line through the PCR data set. From the slope of this line the PCR efficiency of each individual sample is calculated. The mean PCR efficiency per amplicon and the Ct value per sample are used to calculate a starting concentration per sample,expressed in arbitrary fluorescence units. Data input and output are through an Excel spreadsheet.
只需要混样,不需要做梯度
步骤: (以伯乐CFX96为例,不太清楚ABI的机器)
实验:为标准qPCR实验。
qPCR 数据输出 : LinRegPCR 可以识别两种形式的输出文件形式:RDML或者quantification Amplification result.实际上就是机器对循环数和荧光信号的实时检测值,通过对线性区段的荧光变化值分析得出扩增效率。
数据选择:理论上RDML值应该是可以用的,估计是我电脑的问题软件并不能识别RDML,所以我已excel输出值作为原始数据,建议先对数据进行粗筛选,例如加样失败的等造成的点可以在输出的数据中删掉(当然不删也可以,LinRegPCR后期会对这些点进行忽略)
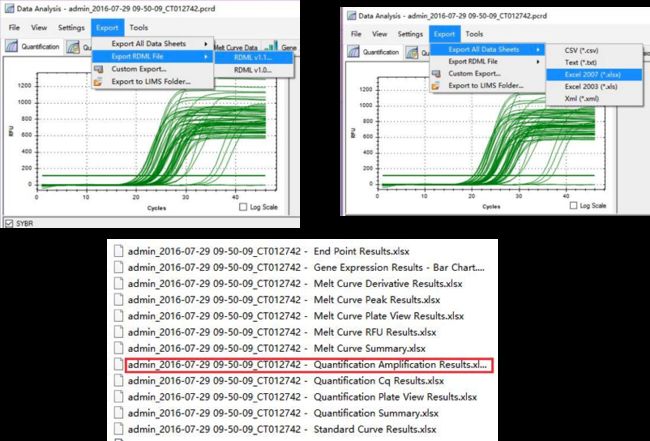

数据输入:打开 qualification amplification results. xls, →打开 LinRegPCR →文件→ read from excel → 如图7选择参数→ OK → 点击determine baselines
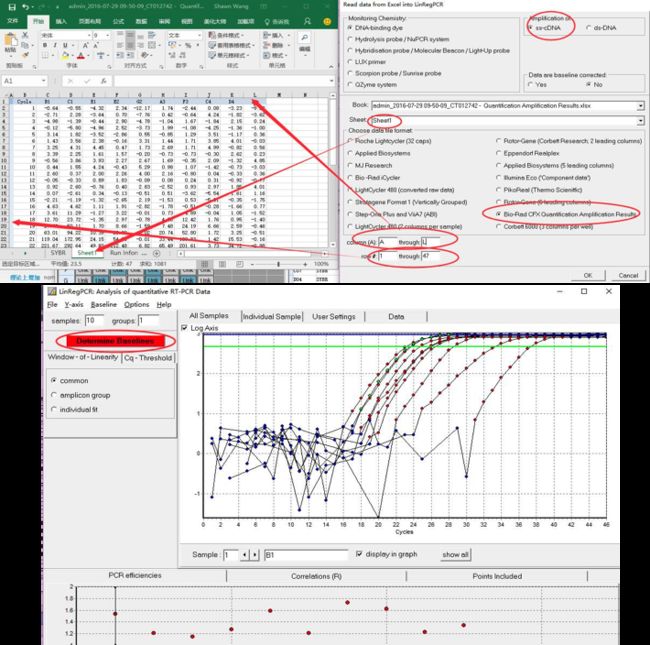
结果:如果没有做重复则不需要分组,如果做了重复在sample grouping中可以编辑分组,indentifier 中键入基因的名称,然后相同的会自动分为一组。最后,点击文件,输出excel,查看结果,每个孔的扩增效率及R2结果都会显示,其次如果你分了组,会显示校正过的扩增效率均值。保证每个引物的扩增效率位于85%-115%之间,过大或者过小都说明引物扩增效率较差。

实验过程:
RNA的质量要求:
纯度:1.7 <OD260/OD280<2.0(<1.7时表明有蛋白质或酚污染;>2.0时表明可能有异硫氰酸残存。 干净的核酸A260/A230应该在2左右。如果有在230 nm 处的强吸收提示污染有酚盐离子等有机化合物。具体吸光值和纯度的关系看这篇文章:OD260/OD280比值的问题-丁香通;此外可以用1.5%的琼脂糖凝胶电泳检测,具体方法:RNA琼脂糖凝胶电泳-丁香通,要注意的是如果只检测纯度是否准确用非变性胶检测,不要点marker,因为ssRNA没有变性的情况下RNA的涌动率和分子量对数不程线性关系,无法正确表示分子量。
浓度:理论上不低于100ng/ul,如果浓度太低,一般情况下纯度也不高

此外,如果样品比较珍贵,且RNA浓度较高时建议提完后可以分装,将RNA稀释到终浓度100-300ng/ul做反转录。
反转录过程
在进行mRNA转录时,采用可以特异结合到polyA尾巴的oligo(dt)引物反转录,而lncRNA,circRNA则采用随即六聚体(Random 6 mer)引物对总RNA进行反转录,对于miRNA则使用miRNA特异的颈环引物进行反转录,很多公司现在推出了专门的加尾法试剂盒,对于茎环法而言,加尾法更加便捷,高通量,节省试剂,但是对于同家族的miRNA区分效果应该没有茎环法好。对于基因特异性引物(茎环)的浓度各反转录试剂盒都有要求。miRNA现在用的内参为U6,在茎环法反转的过程中要单独反转录一管U6,直接加入U6的前后引物。而circRNA和lncRNA均可以用HKGs作为内参。
cDNA检测
一般情况下RNA如果没有问题,cDNA也应该没有问题,但如果追求实验的完美性,最好用一个可以区分gDNA和cds的内参基因(Reference gene,RG)一般RG为管家基因(housekeeping gene, HKG)如图10;当时在做大豆贮藏蛋白,用了包含内含子的actin7作为内参,该引物在gDNA中的扩增片段大小为452bp,cDNA为模板的话为142bp,那么检测结果发现有部分cDNA其实受到了gDNA的污染,同时也证明了反转录的结果没有问题,完全可以作为PCR的模板。直接用cDNA跑琼脂糖凝胶电泳没有用,跑出来是弥散的带,说服力不强。

qPCR条件的确定
一般来说按照试剂盒的protocol来没有问题,主要是在tm值这一步,如果在引物设计时部分引物非常设计,导致tm值与理论的60℃上下差异较大,建议将cDNA样品混合后用引物跑一个梯度PCR,尽量避免将没有条带的温度设为TM值。
数据分析
常规的相对荧光定量PCR的处理方法基本是按照2-ΔΔCT.数据处理模板。