上期我们做了肝癌组织399个miRNA样本的 肿瘤 VS 正常 的差异表达分析.但样本质量控制时我们发现样本差距过大,生物变异系数过高.部分正常组织和肿瘤组织聚在一起,本期我们对这399个样本进一步分析.
先将样本信息从头梳理一遍,TCGA肝癌miRNA病例(cases)共有373个,但是却有425个表达谱文件样本,说明其中有部分病例做了重复(可能重复两次也可能重复三次,这个无所谓).之前我们先根据annotations.txt文件去除了23个异常样本文件,剩下了402个样本.然后在做差异表达分析时发现这402个样本中有43个正常(Solid Tissue Normal),3个复发(Recurrent Tumor)和356个肿瘤(Primary Tumor),我们又把3个复发的去掉了.现在剩下399个样本(356 Tumor和43 Normal,并且这43个Normal是来源于356 Tumor中43个cases的癌旁正常组织),然后使用edgeR做了356 Tumor VS 43 Normal的差异表达分析.
在上期的分析中我们发现Tumor样本和Normal样本并没有各自聚集在一起,而是大量Tumor和Normal聚集在了一起,如下图:
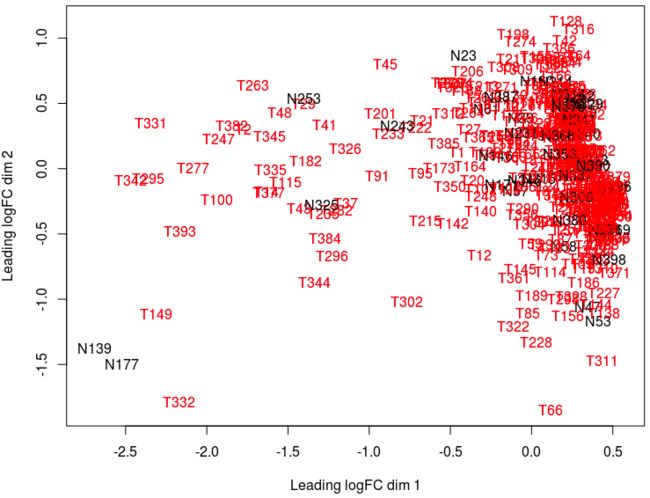
我们先对这399个样本聚类看看,先读入文件,和上期的一样:
rm(list=ls())
library(data.table)
library(edgeR)
library(stringi)
# 表达谱文件路劲:
miRNA_exp_path="./TCGA_liver_miRNA/exp_matrix_reads.tsv"
# 样本信息+临床信息文件路径:
cases_annotation_path="./TCGA_liver_miRNA/cases_files_merged_survival.tsv"
# 使用data.table的fread读入表达谱文件(reads count)
miRNA_exp<-fread(miRNA_exp_path,showProgress = T,header = T,sep = "\t")
# 去掉最后一列空行,如果是read.table则不会有这个问题
names(miRNA_exp)[ncol(miRNA_exp)]<-"final_col"
miRNA_exp<-miRNA_exp[,!c("final_col")]
# 使用data.table的fread读入样本说明信息
cases_annot<-fread(cases_annotation_path,showProgress = T,header = T)
# 去掉3个复发的,在表达量矩阵中去掉:
grp<-as.factor(merge(data.table(file_id=names(miRNA_exp)),cases_annot[,c("file_id","case_id","cases_0_samples_0_sample_type")],by="file_id")[,cases_0_samples_0_sample_type])
recurrent_tumor_id<-merge(data.table(file_id=names(miRNA_exp)),cases_annot[,c("file_id","case_id","cases_0_samples_0_sample_type")],by="file_id")[,file_id][which(grp=="Recurrent Tumor")]
miRNA_exp<-miRNA_exp[,-recurrent_tumor_id,with=F]
# 重新获得样本信息:
grp<-as.factor(merge(data.table(file_id=names(miRNA_exp)),cases_annot[,c("file_id","case_id","cases_0_samples_0_sample_type")],by="file_id")[,cases_0_samples_0_sample_type])
# 查看样本信息:
table(grp)
# 保存miRNA名称为列名
miRNA_names<-miRNA_exp[,miRNA]
miRNA_exp<-miRNA_exp[,!c("miRNA")]
# 将data.table转为data.frame,因为data.table没有行名,前面使用data.table是因为速度快
setDF(miRNA_exp)
# 添加列名
rownames(miRNA_exp)<-miRNA_names
# 拷贝个新的
miRNA_exp_2<-copy(miRNA_exp)
# 使用grp+列数修改列名
grp_chr<-as.character(grp)
grp_chr[which(grp_chr=="Primary Tumor")]<-"T"
grp_chr[which(grp_chr=="Solid Tissue Normal")]<-"N"
grp<-as.factor(grp_chr)
colnames(miRNA_exp_2)<-paste(grp,1:length(grp),sep="")
进行聚类:
# 进行系统聚类
d<-dist(t(miRNA_exp_2))
hc1<-hclust(d)
plot(hc1,cex=0.4,hang=-1)
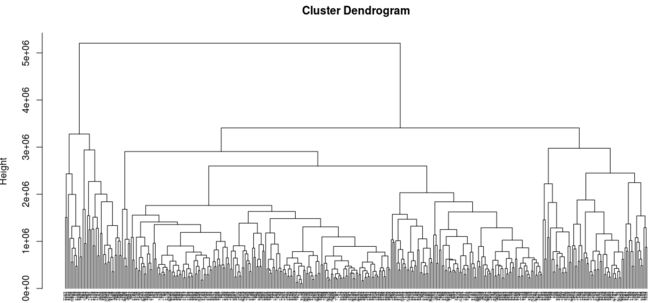
由于样本比较多有些看不清,但放大后发现Tumor和Normal还是没有各自聚在一起,混杂在一起毫无规律.
用Kmeans聚类看看能不能将Tumor和Normal的样本分开:
# kmeans聚类
km<-kmeans(t(miRNA_exp_2),2)
km_cluster<-as.data.frame(km$cluster)
# Normal里的分类:
table(km_cluster[which(substr(rownames(km_cluster),1,1)=="N"),])
# Tumor里的分类:
table(km_cluster[which(substr(rownames(km_cluster),1,1)=="T"),])
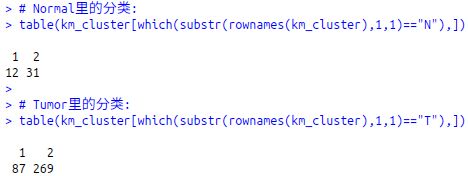
Kmeans聚类结果和已知的Tumor/Normal样本分类相比较发现还是没能很好的聚在一起.也就是说按照现有的表达谱没法区分样本类型.
其实这399个样本可以分为3个部分:(一)43个正常样本,(二)与43个正常样本同一case的肿瘤样本,(三)313个其他肿瘤样本.我们猜Tumor/Normal样本混杂在一起的原因可能是由于是因为来源于同一case导致的.
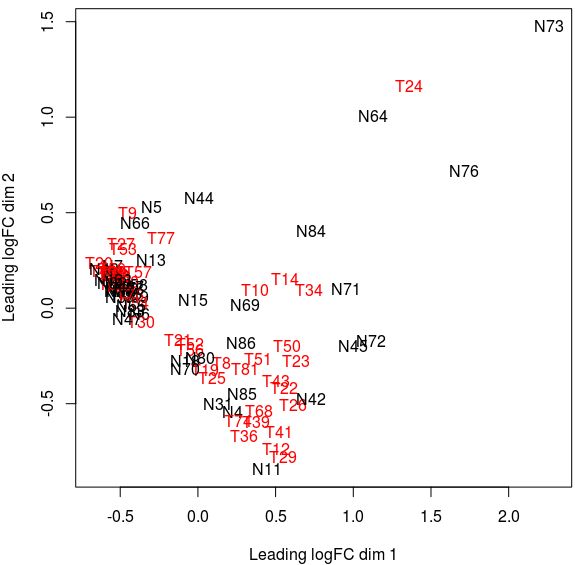
我们先看看43个正常样本和)与43个正常样本同一case的肿瘤样本的聚集信息:
# 找出Normal样本所对应的同一case的Tumor样本:
cases_annot_sub<-cases_annot[c(colnames(miRNA_exp)),on="file_id"]
normal_file_id<-cases_annot_sub[cases_0_samples_0_sample_type=="Solid Tissue Normal",c("file_id","case_id")]
cases_N_T<-cases_annot_sub[normal_file_id$case_id,on="case_id"]
miRNA_exp_NT<-miRNA_exp[,which(names(miRNA_exp) %in% cases_N_T$file_id)]
grp<-as.factor(merge(data.table(file_id=names(miRNA_exp_NT)),cases_annot[,c("file_id","case_id","cases_0_samples_0_sample_type")],by="file_id")[,cases_0_samples_0_sample_type])
# 使用grp+列数修改列名
grp_chr<-as.character(grp)
grp_chr[which(grp_chr=="Primary Tumor")]<-"T"
grp_chr[which(grp_chr=="Solid Tissue Normal")]<-"N"
grp<-as.factor(grp_chr)
colnames(miRNA_exp_NT)<-paste(grp,1:length(grp),sep="")
# 建立一个DGEList对象,并对数据进行标准化(normalize)
d <- DGEList(counts=miRNA_exp_NT, group=grp)
# 使用TMM方法归一化:
d <- calcNormFactors(d,method = "TMM")
head(d$samples)
cols <- as.numeric(d$samples$group)
plotMDS(d,col=cols)
再看313非同一case的tumor和43 normal的样本聚集情况
# 找出Normal样本所对应的不同case的Tumor样本:
cases_annot_sub<-cases_annot[c(colnames(miRNA_exp)),on="file_id"]
normal_file_id<-cases_annot_sub[cases_0_samples_0_sample_type=="Solid Tissue Normal",c("file_id","case_id")]
tumor_in_normal_file_id<-cases_annot_sub[normal_file_id$case_id,on="case_id"][cases_0_samples_0_sample_type=="Primary Tumor"][,c("case_id","file_id")]
miRNA_exp_NT<-miRNA_exp[,which(!names(miRNA_exp) %in% tumor_in_normal_file_id$file_id)]
grp<-as.factor(merge(data.table(file_id=names(miRNA_exp_NT)),cases_annot[,c("file_id","case_id","cases_0_samples_0_sample_type")],by="file_id")[,cases_0_samples_0_sample_type])
# 使用grp+列数修改列名
grp_chr<-as.character(grp)
grp_chr[which(grp_chr=="Primary Tumor")]<-"T"
grp_chr[which(grp_chr=="Solid Tissue Normal")]<-"N"
grp<-as.factor(grp_chr)
colnames(miRNA_exp_NT)<-paste(grp,1:length(grp),sep="")
# 建立一个DGEList对象,并对数据进行标准化(normalize)
d <- DGEList(counts=miRNA_exp_NT, group=grp)
# 使用TMM方法归一化:
d <- calcNormFactors(d,method = "TMM")
head(d$samples)
cols <- as.numeric(d$samples$group)
plotMDS(d,col=cols)
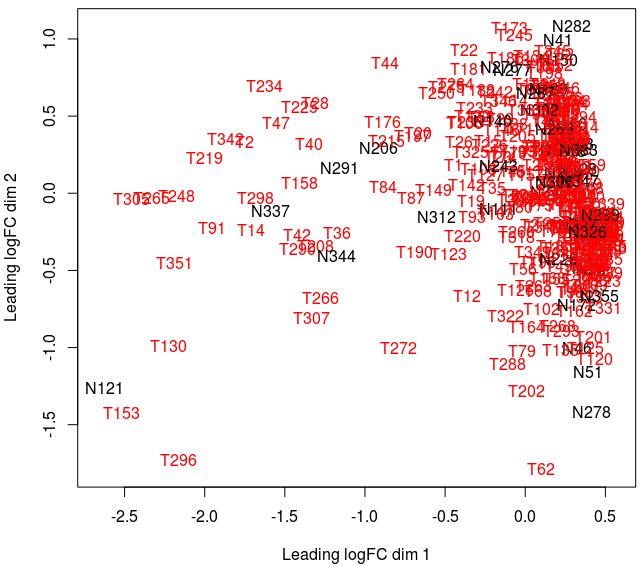
同一case的tumor和normal(图四)样本聚在一起分不开(左边基本聚到一点了),图五非同一case的还是非常杂乱,先不管了,直接忘下做差异表达分析了.代码和上期的一样就不放了,加上昨天的一共有三个差异表达分析结果:356 Tumor vs 43 Normal的;43 Tumor vs 43 Normal的;313 Tumor vs 42 Normal的.结果发现这三次的差异表达结果基本没有交集,甚至有些miRNA的上下调都是反的,部分结果如下:
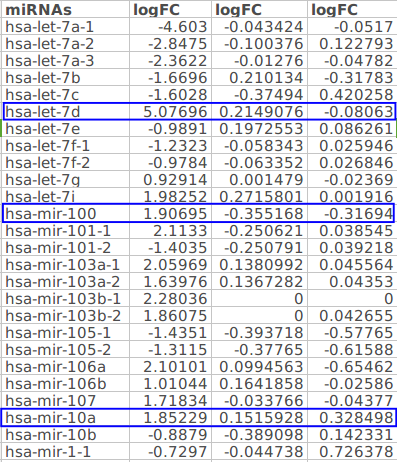
只看logFC部分都差的不是一星半点,我随便看了下蓝线框起来的部分,如hsa-let-7d在dbDEM数据库(一个miRNA差异表达数据库)的结果:
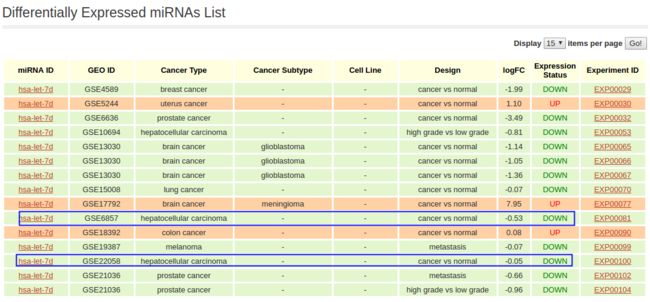
在dbDEMC数据库中let-7d都下调,说明第三次结果即313 vs 43可能是更准确的.
还有hsa-mir-100的:
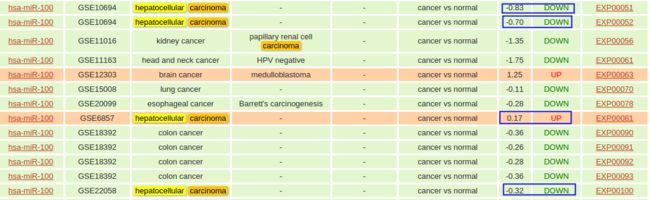
在dbDEMC数据库中mir-100三下调一上调,说明第二(43 vs 43)和三次结果(313 vs 43)可能是更准确的.
再看下hsa-mir-10a的:
在dbDEMC数据库中mir-10a两个结果都下调,但由TCGA得到的都是上调.说实话我有点方...
现在我非常怀疑从肿瘤病人身上获得的正常组织可不可以拿来做差异表达分析的对照组.一是由于之前的聚类分析表明这两大类(Tumor/Normal)样本基本没能分开,就是这两类表达量总体来看区别不大.二是由于三次差异表达分析结果相差甚远.第一次相当于总体分析,第二三次相当于抽样分析,没管p值,只看logFC都有这么大的差异.说明同一case的肿瘤样本和正常样本对于差异表达分析结果有很大影响.
为了排除这种影响,下期我将使用过GTEx的正常样本而不是TCGA的正常样本作为对照组进行差异表达分析.
更多原创精彩内容敬请关注生信杂谈:
