宏基因组分析教程
microPITA | 宏基因组测序前,你可以这样筛选样本
microPITA
加拿大安大略研究所建立的生物信息网
1. 什么是 组装?
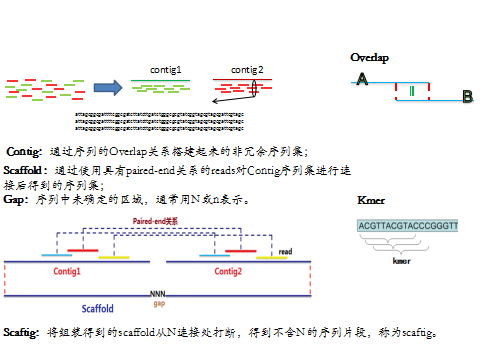
基因组测序时将测得的各短序列拼接成连续完整的序列
简单地说就是从reads 到 Scaftig的过程。
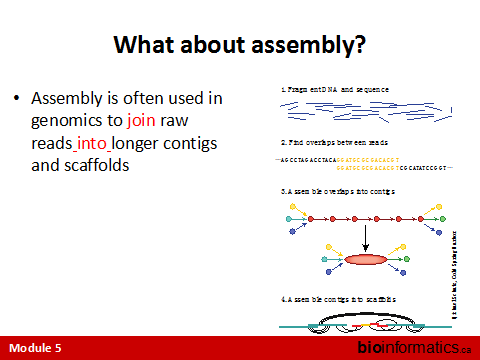
2. 为什么要组装?
因为目前二代测序的序列读长比较短最长只有300bp
- Assembly improves annotation accuracy

3. 怎样组装?
经过预处理后得到 Clean Data,使用 SOAP denovo(肠道样品用soapdenovo || soil,water用MEGAHIT)组装软件进行组装分析( Assembly Analysis )
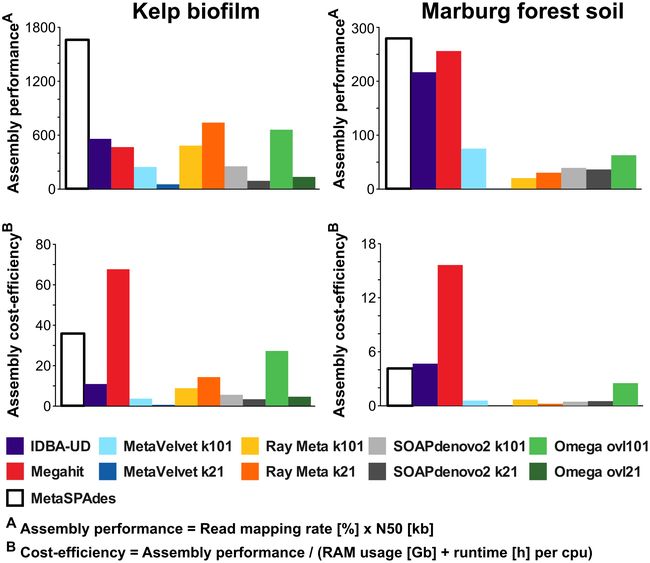
如何选择组装软件:↓↓↓
if (micro diversity is not a major issue&& the primary research goal is to bin && reconstruct representative bacterial genomes from a given environment){
metaSPAdes should clearly be the assembler of choice. # This assembler yields the best contig size statistics while capturing a high degree of community diversity, even at high complexity and low read coverage;
}elsif(mico diversity is however an issue || the degree of
captured diversity is far more important than contig
lengths){
then IDBA-UD or Megahit should be preferred. # The sensitivity of these assemblers, both for diversity as well as micro diversity, makes them optimal choices when trying to discover novel species in complex habitats. Whenever computational resources become limiting,
Megahit becomes the most attractive option, due to its good compromise between contig size statistics, captured diversity and required memory.
}
However, the bias of Megahit towards relatively low coverage genomes may provide a disadvantage for very large datasets, leading to a suboptimal assembly of high abundant community member genomes.
In such cases, Megahit may provide better results when assembling subsets of the sequencing data in a “divide and conquer” approach.
- Published: January 18, 2017 · plosone
Comparing and Evaluating Metagenome Assembly Tools from a Microbiologist’s Perspective - Not Only Size Matters!
宏基因组组装有两种常见策略:
- 1基于序列overlap关系进行拼接,代表软件有Omega;
- 2 基于de Bruijn图进行组装
由于现阶段的主流测序方法是二代短片段测序,序列短而且数目庞大,如果利用overlap关系直接进行组装,这要求每对reads之间都进行一次序列比较,这会很耗费时间,而且结果并不可靠。为迎合二代测序的特点,一种基于k-mer的de Bruijn组装策略则成为更有效的解决方法。

SOAPdenovo-63mer all -d 1 -M 3 -R -u -F -s KB1.soapdenovo.cfg -K 55 -o 55 1>ass.55.log 2>ass.55.err
-d 去除kmers频数不大于该值(kmerFreqCutoff)的k-mer,默认值[0] ##最小化错误测序带来的影响
-M 在contiging操作时,合并相似序列的强度,默认值为[1],最小值0,最大值3。#deal with heterozygosis
-R (optional) 移除repeats,使用pregraph步骤中产生的结果,如果参数-R在pregraph步骤中被设置的话,默认[NO]
-u (optional) 构建scaffolding前不屏蔽高/低覆盖度的contigs,这里高频率覆盖度指平均contig覆盖深度的2倍。默认[mask]屏蔽
-F (optional) 对scaffold内部的gap进行填充,这个参数现在似乎没什么用,因为SOAPdenovo附带了一个Gapcloser工具,就是用于scaffold内部填充的。
-s solexa reads 的配置文件
-K 输入的K-mer值大小,默认值[23],取值范围 13-127 #K-mer值必须是奇数;组装杂合子基因组的K-mer值应该小一点;组装含有高repeats基因组且要求其有高的测序深度和长的reads,的K-mer应该大一点。
-o 图形输出的文件名前缀
k-mer 如何影响宏基因组组装 ?
使用de Bruijn graph组装基因组的时候,Kmer数为何必须是奇数呢
算法:SOAPdenovo的一个组装过程
SOAPdenovo组装软件使用记录

目前最好最完整的SOAPdenovo使用说明
基因组组装工具之 SOAPdenovo 使用方法
基因组组装
组装结果评价
-
N50(N90)的定义
指基因组组装结果中,一半的scaffolds/ contigs长度都大于这个值。
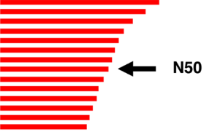 N50
N50
1、 序列一致性评估:
2、 序列完整性评估:
3、 准确性评估:
4、 保守性基因评估:
基因组组装效果评估
轻松get干货—《基因组注释与基因注释原理及常用软件使用方法》
混合组装
什么是混合组装
将各样品未被利用上的 reads 放在一起进行组装
为什么要混合组装
以期发现样品中的低丰度物种信息
考虑到在宏基因组组装中reads利用率很低,单样品5Gb测序量情况下,环境样品组装reads利用率一般只有10%左右,肠道样品或极端环境样品组装reads利用率一般能达到30%
怎样进行混合组装
- Reads mapping
What is a read mapping?
Reads_mapping 来找出上一步单样本未被利用的reads
bowtie2-build --large-index B11.2.scaftigs.fa B11.2.scaftigs.fa 2> bwt.log
bowtie2 --end-to-end --sensitive -I 200 -X 400 --threads 8 -x KB2.scaftigs.fa -1 KB2_350.nohost.fq1.gz -2 KB2_350.nohost.fq2.gz -S KB2_350.bowtie.sam 2> bowtie.log
-x 由bowtie2-build所生成的索引文件的前缀。首先 在当前目录搜寻,然后在环境变量 BOWTIE2_INDEXES 中制定的文件夹中搜寻。
-1 双末端测寻对应的文件1。可以为多个文件,并用逗号分开;多个文件必须和 -2 中制定的文件一一对应。比如:"-1 flyA_1.fq,flyB_1.fq -2 flyA_2.fq,flyB_2.fq". 测序文件中的reads的长度可以不一样。
-2 双末端测寻对应的文件2.
-U 非双末端测寻对应的文件。可以为多个文件,并用逗号分开。测序文件中的reads的长度可以不一样。
-S 所生成的SAM格式的文件前缀。默认是输入到标准输出。
↑↑↑↑↑↑↑必须参数↑↑↑↑↑↑↑↑↓↓↓↓↓↓↓↓↓↓↓↓可选参数:↓↓↓↓↓↓↓↓↓↓↓↓↓↓
--end-to-end 比对是将整个read和参考序列进行比对. 该模式--ma的值为0. 该模式为默认模式
--sensitive Same as: -D 15 -R 2 -N 0 -L 22 -i S,1,1.15 (default in --end-to-end mode)
-I/--minins 设定最小的插入片段长度. Default: 0.
-X/--maxins 设定最长的插入片段长度. Default: 500.
-p/--threads NTHREADS 设置线程数. Default: 1
Bowtie2使用方法与参数详细介绍
-
unmmaped.assembly
SOAPdenovo-63mer all -d 1 -M 3 -R -u -F -s NOVO_MIX.soapdenovo.cfg -K 55 -o 55 1>ass.55.log 2>ass.55.err
句句干货!一文读懂宏基因组binning
Microbiome Helper: a Custom and Streamlined Workflow for Microbiome Research
rrnDB: Stoddard et al