为达到更好的浏览效果及查看全部图片,可转至RPubs
1 安装一些R包
数据包: ALL, CLL, pasilla, airway
软件包:limma,DESeq2,clusterProfiler
工具包:reshape2
绘图包:ggplot2
rm(list = ls())
if(F){
source("http://bioconductor.org/biocLite.R")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
options(BioC_mirror="http://mirrors.ustc.edu.cn/bioc/")#修改镜像,安装会加速
BiocManager::install("clusterProfiler")
BiocManager::install("ComplexHeatmap")
BiocManager::install("maftools")
BiocManager::install("reshape2")
}
2.了解ExpressionSet对象
比如CLL包里面就有data(sCLLex) 找到它包含的元素,提取其表达矩阵(使用exprs函数),查看其大小
A.参考:http://www.bio-info-trainee.com/bioconductor_China/software/limma.html
B.参考:https://github.com/bioconductor-china/basic/blob/master/ExpressionSet.md
library(CLL)
data("sCLLex")
View(sCLLex)#查看其包含的元素
sCLLex#查看包含的元素
exp <- exprs(sCLLex)#提取表达矩阵
dim(exp)#查看其大小
group <- pData(sCLLex)#提取分组信息
table(group)#设计矩阵
3了解 str,head,help函数
作用于 第二步提取到的表达矩阵
str(exp)
head(exp)
help(exp)
4安装并了解 hgu95av2.db 包
看看 ls("package:hgu95av2.db") 后 显示的那些变量
library(hgu95av2.db)
ls("package:hgu95av2.db")
ls(hgu95av2.db)
5理解 head(toTable(hgu95av2SYMBOL)) 的用法
找到 TP53 基因对应的探针ID
head(toTable(hgu95av2SYMBOL))
ids <- toTable(hgu95av2SYMBOL)
以下几种方法获取TP53的探针名称
1 subset函数
a1 <- subset(ids, ids$symbol=='TP53')
2 grep函数
a4 <- ids[grep("^TP53$", ids$symbol),]
3 filter函数(依赖dplyr包)
a5 <- filter(ids, ids$symbol=='TP53')
6 理解探针与基因的对应关系
总共多少个基因,基因最多对应多少个探针,
是哪些基因,是不是因为这些基因很长,所以在其上面设计多个探针呢?总共多少gene
length(unique(ids$symbol))
对应探针多的是那些gene
ids_fre <- data.frame(table(ids$symbol))
tail(ids_fre[order(ids_fre$Freq),])
或者用sort命令
tail(sort(table(ids$symbol)))
不管是Agilent芯片,还是Affymetrix芯片,上面设计的探针都非常短。
最长的如Agilent芯片上的探针,往往都是60bp,但是往往一个基因的长
度都好几Kb。因此一般多个探针对应一个基因,取最大表达值探针来作为
基因的表达量。
7.找到芯片有而hgu95av2.db中没有对应基因名的探针
第二步提取到的表达矩阵是12625个探针在22个样本的表达量矩阵,
找到那些不在 hgu95av2.db 包收录的对应着SYMBOL的探针。
提示:有1165个探针是没有对应基因名字的。
第一种方法:match函数
dim(exp)
dim(hgu95av2SYMBOL)
b1 <- exp[-match(ids$probe_id, rownames(exp)),]
dim(b1)
第二种方法:%in%函数
b2 <- exp[!(rownames(exp) %in% ids$probe_id),]
b1==b2
8 过滤表达矩阵:删除注释包中没有对应基因名的探针
过滤表达矩阵,删除那1165个没有对应基因名字的探针。
exp_filter_no_symbol <- as.data.frame(exp[match(ids$probe_id, rownames(exp)),])
exp_filter_no_symbol2 <- as.data.frame(exp[rownames(exp) %in% ids$probe_id,])
9 整合表达矩阵,多个探针到一个探针
多个探针对应一个基因的情况下,只保留在所有样本里面平均表达量最大的那个探针。
提示,理解 tapply,by,aggregate,split 函数 , 首先对每个基因找到最大表达量的探针。
然后根据得到探针去过滤原始表达矩阵
这种方法能够一定程度上增加差异基因的数目,但也容易造成假阳性的结果
https://blog.csdn.net/tommyhechina/article/details/80468361
合并探针,先看有无NA值,若有,可以删除或填充
先检查有无,若返回值>0,说明有NA值。
length(which(is.na(exp_filter_no_symbol)))
结果是0,说明没有NA值。如果有则用impute包来进行填充
exp_symbol<- impute.knn(exp_symbol)$data
下面开始进行合并,用aggregate命令
ids <- toTable(hgu95av2SYMBOL)
ids$median <- apply(exp_filter_no_symbol, 1, median)
ids <- ids[order(ids$symbol, ids$median, decreasing = T),]
ids_filtered <- ids[!duplicated(ids$symbol),]
dim(ids_filtered)
10 过滤后的表达矩阵行名改为SYMBOL
把过滤后的表达矩阵更改行名为基因的symbol,因为这个时候探针和基因是一对一关系了。
exp_filter_no_symbol$probe_id <- rownames(exp_filter_no_symbol)
exp_ids <- merge(ids_filtered, exp_filter_no_symbol, by ='probe_id' )
rownames(exp_ids) <- exp_ids$symbol
exp_sym <- exp_ids[,-c(1:3)]
11 画第一个样本和所有样本的基于表达量的图
对第10步得到的表达矩阵进行探索,先画第一个样本的所有基因的表达量的boxplot,hist,density , 然后画所有样本的 这些图
参考:http://bio-info-trainee.com/tmp/basic_visualization_for_expression_matrix.html
理解ggplot2的绘图语法,数据和图形元素的映射关系
boxplot(exp_sym$CLL11.CEL)

boxplot(exp_sym)
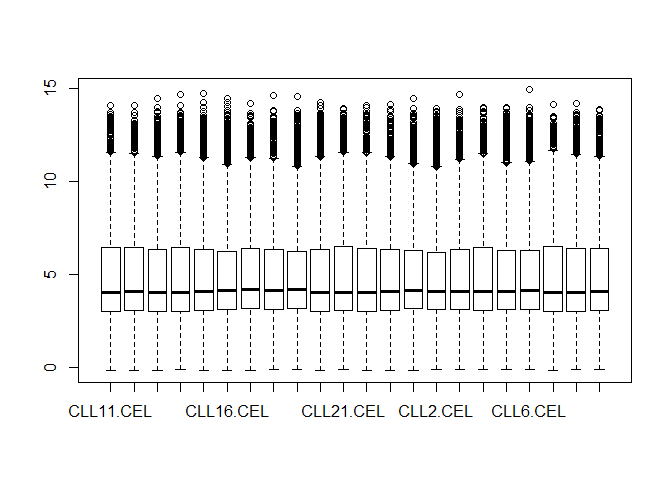
用ggplot2来画
ggplot2画图要改变数据形式
library(reshape2)
exp_g <- melt(exp_ids[,-c(1,3)], id.vars = 'symbol')
exp_g$group <- rep(group$Disease, each = nrow(exp_sym))
colnames(exp_g)[2] <- 'sample'
head(exp_g)
第一个样本的
library(ggplot2)
sample_1 <- subset(exp_g, exp_g$sample=='CLL2.CEL')
head(sample_1)
p_s1 <- ggplot(sample_1, aes(x = sample, y = value, fill = 'lightblue'))
p_s1 + geom_boxplot()
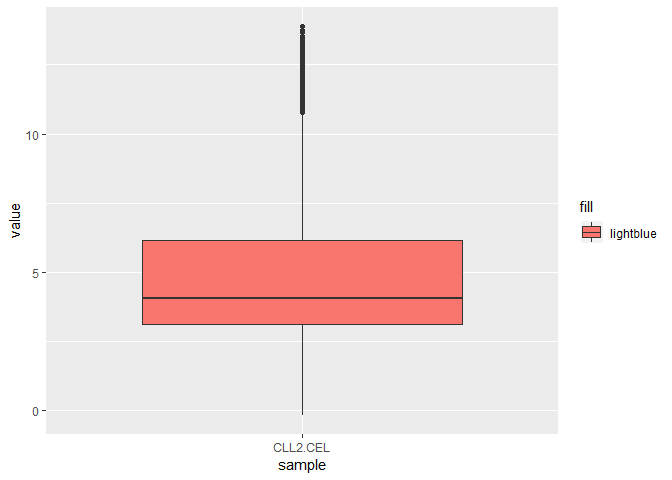
p_s1 + geom_violin()+geom_boxplot(fill = 'white')

ggplot(sample_1, aes(x = value, fill = 'lightblue'))+
geom_histogram(bins = 500)

ggplot(sample_1, aes(x = value, fill = 'lightblue'))+
geom_density()

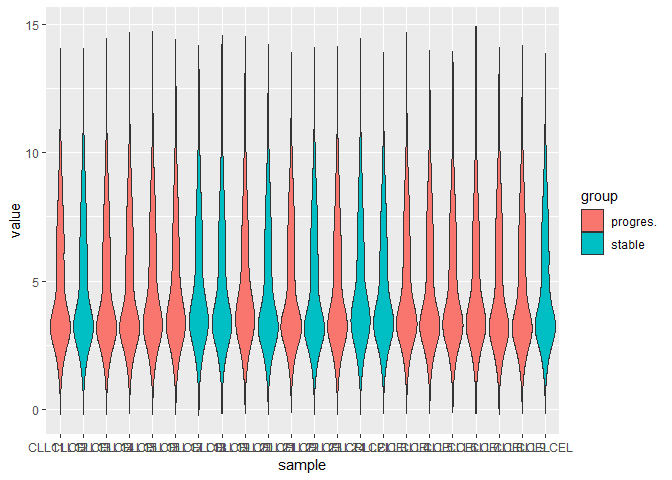
下面是所有样本的
p <- ggplot(exp_g, aes(x = sample, y = value, fill = group))
p + geom_boxplot()

p + geom_violin()
ggplot(exp_g, aes(x=value,fill = group))+ geom_histogram(bins = 500)+
facet_wrap(~group, nrow = 5)

ggplot(exp_g, aes(x=value,fill = group))+ geom_density()+
facet_wrap(~group, nrow = 5)

12 理解统计学指标
mean,median,max,min,sd,var,mad,并计算出每个基因在所有样本的这些统计学指标,最后按照mad值排序,取top 50 mad值的基因,得到列表
head(exp_sym)
exp_mean <- apply(exp_sym, 1, mean)
exp_median <- apply(exp_sym,1, median)
exp_max <- apply(exp_sym, 1, max)
exp_min <- apply(exp_sym, 1, min)
exp_sd <- apply(exp_sym, 1, sd)
exp_var <- apply(exp_sym, 1, var)
exp_md <- apply(exp_sym, 1, mad)
exp_sta <- data.frame(exp_mean, exp_median, exp_max, exp_min, exp_sd, exp_var, exp_md)
md_order <- exp_sta[order(exp_sta$exp_md, decreasing = T),]
md_order[1:50,1:5]
13 提取基因的表达矩阵子集,绘制热图
根据第12步骤得到top 50 mad值的基因列表来取表达矩阵的子集,并且热图可视化子表达矩阵。试试看其它5种热图的包的不同效果。
names_50 <- rownames(md_order[1:50,])
exp_mad_top50 <- exp_sym[match(names_50, rownames(exp_sym)),]
pheatmap::pheatmap(log2(exp_mad_top50))

pheatmap::pheatmap(exp_mad_top50, scale=c("row"))

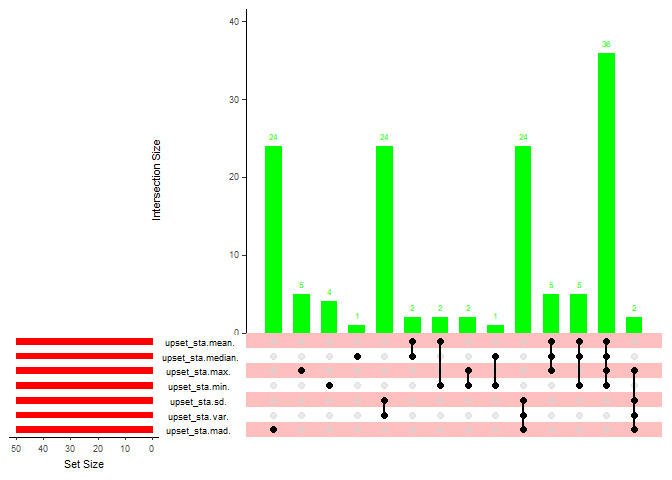
14 取N个指标的交集并可视化:UpSetR包
取不同统计学指标mean,median,max,mean,sd,var,mad的各top50基因列表,
使用UpSetR包来看他们之间的overlap情况
mean_50 <- head(sort(exp_mean, decreasing = T),50)
median_50 <- head(sort(exp_median, decreasing = T),50)
写函数完成上述重复问题问题
sta_50 <- function(x){
x <- head(sort((apply(exp_sym, 1, x)), decreasing = T),50)
x_50 <- x
return(names((x_50)))
}
library('UpSetR')
取不含重复的并集
sta_50_all <- unique(c(sta_50(mean), sta_50(median),sta_50(max),sta_50(min),sta_50(sd),sta_50(var), sta_50(mad)))
转成upset需要的格式,也就是1和0分别代表
u_mean <- ifelse(sta_50_all %in% sta_50(mean), 1, 0)
再写个函数
upset_sta <- function(x){
x <- ifelse(sta_50_all %in% sta_50(x), 1, 0)
u_x <- x
return(u_x)
}
upset_all <- data.frame(sta_50_all, upset_sta(mean),upset_sta(median),
upset_sta(max),upset_sta(min),upset_sta(sd),upset_sta(var),upset_sta(mad))
?upset
upset(upset_all, nsets = 7, matrix.color = 'black',main.bar.color = 'green',
sets.bar.color = 'red',point.size = 2, line.size = 0.8,
shade.color = 'red', matrix.dot.alpha = 0.5)
15 提取表型数据
在第二步的基础上面提取CLL包里面的data(sCLLex) 数据对象的样本的表型数据。
data("sCLLex")
pData(sCLLex)
16 对表达矩阵进行聚类画图,添加临床表型数据
对所有样本的表达矩阵进行聚类并且绘图,然后添加样本的临床表型数据信息(更改样本名)
前面
head(exp_sym)
group_list <- as.character(pData(sCLLex)[,2])
colnames(exp_sym) <- paste(group_list, 1:22, sep = '')
head(exp_sym)
t.exp <- t(exp_sym)
t.exp[1:5,1:5]
hc <- hclust(dist(t.exp))
plot(as.dendrogram(hc))

用factoextra包画
exp_cluster <- t(exp_sym)
exp_clust_dist <- dist(exp_cluster, method = 'euclidean')
hc <- hclust(exp_clust_dist,'ward.D')
library(factoextra)
fviz_dend(hc, k=4, cex = 0.5,
k_colors = c("#2E9FDF", "#00AFBB", "#E7B800", "#FC4E07"),
color_labels_by_k = TRUE, rect = TRUE)

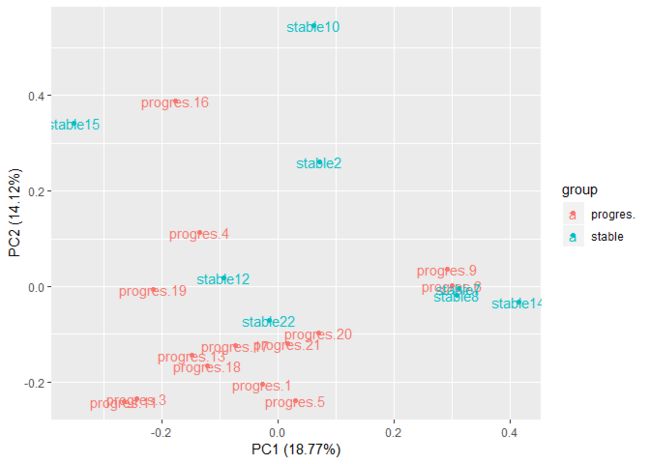
17 所有样本PCA绘图
对所有样本的表达矩阵进行PCA分析并且绘图,同样要添加表型信息
library(devtools)
install_github('sinhrks/ggfortify')
library(ggfortify)
df <- as.data.frame(t(exp_sym))
df$group <- group$Disease
autoplot(prcomp(df[,1:(ncol(df)-1)]), data=df,
label = TRUE, colour = 'group')
18. 批量T检验
根据表达矩阵及样本分组信息进行批量T检验,得到检验结果表格
dat <- exp_sym
group_list <- as.factor(group_list)
group1 <- which(group_list == levels(group_list)[1])
group2 <- which(group_list == levels(group_list)[2])
dat1 <- dat[,group1]
dat2 <- dat[,group2]
dat <- cbind(dat1, dat2)
pvals <- apply(dat, 1, function(x){
t.test(as.numeric(x)~group_list)$p.value
})
head(pvals)
p.adj <- p.adjust(pvals, method = 'BH')
avg_1 <- log2(rowMeans(dat1))
avg_2 <- log2(rowMeans(dat2))
log2FC <- avg_2-avg_1
deg_t.test <- cbind(avg_1, avg_2, log2FC, pvals, p.adj)
deg_t.test <- deg_t.test[order(deg_t.test[,4]),]
head(deg_t.test)
class(deg_t.test)
19.使用limma包筛选差异DEGs
对表达矩阵及样本分组信息进行差异分析,得到差异分析表格,
重点看logFC和P值,画个火山图(就是logFC和-log10(P值)的散点图)。
1 构建设计矩阵design matrix
library(limma)
design <- model.matrix(~0+factor(group_list))
colnames(design) <- levels(factor(group_list))
rownames(design) <- colnames(exp_sym)
design
fit <- lmFit(exp_sym, design)
2 构建比较矩阵
contrast.matrix <- makeContrasts(paste0(unique(group_list),
collapse = '-'), levels = design)
fit2 <- contrasts.fit(fit, contrast.matrix)
fit3 <- eBayes(fit2)
3差异表达矩阵获取
mtx <- topTable(fit3, coef = 1, n=Inf)
DEG_mtx <- na.omit(mtx)
dim(mtx)
dim(DEG_mtx)
head(DEG_mtx)
#write.csv(DEG_mtx,"DEG_mtx.csv",quote = F)
4火山图
第一步,设定阈值,界定上调下调和不表达的基因
DEG <- DEG_mtx
logFC_cutoff <- with(DEG, mean(abs(logFC))+2*sd(abs(logFC)))
DEG$change = as.factor(ifelse(DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff,
ifelse(DEG$logFC > logFC_cutoff ,'UP','DOWN'),'NOT')
)
这是两个
ifelse判断嵌套。先了解ifelse的结构,ifelse(条件,yes,no),如果满足条件,那么返回yes/或者执行yes所处的下一个命令;反之返回no,这里外层的ifelse中DEG$P.Value < 0.05 & abs(DEG$logFC) > logFC_cutoff是判断条件,这个就是看p值和logFC是不是达到了他们设定的阈值【p是0.05,logFC是logFC_cutoff】,如果达到了就进行下一个ifelse,达不到就返回NOT;第二层ifelse也是上来一个条件:DEG$logFC > logFC_cutoff,如果达到了,就返回UP即上调基因,达不到就是下调DOWN,最后将判断结果转位因子型,得到DOWN、UP、NOT的三种因子
第二步,设定火山图标题
this_tile <- paste0('Cutoff for logFC is ',round(logFC_cutoff,3),
'\nThe number of up gene is ',nrow(DEG[DEG$change =='UP',]) ,
'\nThe number of down gene is ',nrow(DEG[DEG$change =='DOWN',])
)
第三步,画图
g = ggplot(data=DEG, aes(x=logFC, y=-log10(P.Value), color=change)) +
geom_point(alpha=0.4, size=1.75) +
theme_set(theme_set(theme_bw(base_size=20)))+
xlab("log2 fold change") + ylab("-log10 p-value") +
ggtitle( this_tile ) + theme(plot.title = element_text(size=15,hjust = 0.5))+
scale_colour_manual(values = c('blue','black','red')) ## corresponding to the levels(res$change)
print(g)

第四步,画个漂亮的图
P_volcano=ggplot(DEG,aes(x=logFC,y=-log10(P.Value)))+
geom_point(aes(color=change))+
#设置点的颜色
scale_color_manual(values =c("UP" = "red", "DOWN" = "blue", "NOT" = "grey"))+
labs(x="log2FC",y="-log10FDR")+
#增加阈值线:分别对应FDR=0.05,|log2FC|=1
geom_hline(yintercept=-log10(0.05),linetype=4)+
geom_vline(xintercept=c(-1,1),linetype=4)+
xlim(-3,3)+
theme(plot.title = element_text(size = 25,face = "bold", vjust = 0.5, hjust = 0.5),
legend.title = element_blank(),
legend.text = element_text(size = 18, face = "bold"),
legend.position = 'right',
legend.key.size=unit(0.8,'cm'),
axis.ticks.x=element_blank(),
axis.text.x=element_text(size = 15,face = "bold", vjust = 0.5, hjust = 0.5),
axis.text.y=element_text(size = 15,face = "bold", vjust = 0.5, hjust = 0.5),
axis.title.x = element_text(size = 20,face = "bold", vjust = 0.5, hjust = 0.5),
axis.title.y = element_text(size = 20,face = "bold", vjust = 0.5, hjust = 0.5),
panel.background = element_rect(fill = "transparent",colour = "black"),
panel.grid.minor = element_blank(),
panel.grid.major = element_blank(),
plot.background = element_rect(fill = "transparent",colour = "black"))
P_volcano

20. 比较T检验和limma结果
对T检验结果的P值和limma包差异分析的P值画散点图,看看哪些基因相差很大
deg_t.test <- as.data.frame(deg_t.test)
t_limma_pval <- cbind(deg_t.test,DEG)[,c(4,9)]
head(t_limma_pval)
plot(t_limma_pval)

pheatmap::pheatmap(cor(t_limma_pval))
