RNA-seq数据下载
数据来自文献:An RNA-Seq transcriptome and splicing database of neurons, glia, and vascular cells of the cerebral cortex,GEO编号GSE52564。用Aspera下载原始数据:
# -l设置大些速度更快,默认大概10M; -k 1 设置断点续传 -T不加密
for((i=83; i<=99; i++)); do ascp -k 1 -T -l 800M -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/sra/sra-instant/reads/ByStudy/sra/SRP/SRP033/SRP033200/SRR10337$i/SRR10337$i.sra . ; done &
SRA to fasta
# 并行处理
ls SRR*.sra | xargs -i -n1 -P 5 sh -c "fastq-dump --gzip --split-3 {} && rm {}" &
另外github上有支持多线程的pfastq-dump。需要先安装sratoolkit,可执行的fastq-dump和sra-stat在path中。
pfastq-dump -t 8 -O ./tmp SRR5047994.sra --split-3 --gzip
转录本数据下载
从gencode下载需要的小鼠fasta文件Transcript sequences。当然也可以从Ensemble或者UCSC下载。gencode的转录本有很多预测的,建议用gencode basic子集。本次直接用的UCSC的refMrna.fa。
Kallisto 可以直接用来处理原始的reads
You can run kallisto directly on your (untrimmed) reads.
--Lior Pachter
Kallisto定量处理
index
kallisto index -i refMrna.idx refMrna.fa
quant
# -t 核数 -b 自助法的样本数,作者推荐至少设为30
# 如果下游不需要用booststraps值(比如不做sleuth、QTL分析),
# -b 可以设为0,加快运行速度
for ((i=83;i<=99;i++)); do kallisto quant -i ./ref/mouse_tr/refMrna.idx -o SRR10337$i -t 8 -b 100 SRR10337${i}_1.fastq.gz SRR10337${i}_2.fastq.gz; done &
# 或者如下,单独留个核解压缩
for ((i=83;i<=99;i++)); do kallisto quant -i ./ref/mouse_tr/refMrna.idx -o SRR10337$i -t 7 -b 100 <(zcat SRR10337${i}_1.fastq.gz) <(zcat SRR10337${i}_2.fastq.gz); done &
另外,对于单端测序的命令为
kallisto quant -i transcripts.idx -o output -b 100 --single -l 180 -s 20 reads_1.fastq.gz
除了--single外,还必须给出与建库片段长度(不是read的长度)有关的两个参数:l和s。(典型的Illumina文库的片段长度为180-200bp)
180/20 is a reasonable insert size/std dev estimate for TruSeq RNA v2 library preps
-l 指定建库用的片段长度 -s 建库片段长度的标准差
双端测序会自动估计这两个参数,不需要提供。(官方推荐程序自动估计)
Sleuth差异分析
suppressMessages(library(sleuth))
rm(list = ls())
options(mc.cores = 6L)
### 构造kallisto输出文件路径
setwd(file.path("E:/0ngs","output"))
sample_id <- list.files(pattern = "SRR*")
kal_dirs <- file.path("E:/0ngs","output", sample_id)
### 样品设计分组数据准备
s2c <- read.table(file.path("..","SraRunTable.txt"),header = T,stringsAsFactors = F,sep = "\t")
s2c <- dplyr::select(s2c, sample = Run_s, condition = cell_type_s) # machine = Machine_s
s2c <- dplyr::mutate(s2c, path = kal_dirs)
# 从NCBI下载再转换,注释更准确
# ascp -k 1 -T -l 800M -i ~/.aspera/connect/etc/asperaweb_id_dsa.openssh [email protected]:/gene/DATA/gene2refseq.gz .
# gene <- read.delim("gene.txt",stringsAsFactors = F)
# gene$target_id <- gsub("\\.\\d+","",gene$target_id,perl=T)
# gene <- unique(gene)
# t2g <- gene[gene$target_id %in% transcrip_id,c(3,4,2)]
# t2g <- t2g[order(t2g$Symbol),] #gene排序
# write.table(t2g,"tx2gene.txt",sep = "\t",row.names = F,quote = F)
### 转录本到对应基因注释文件准备
tx2gene <- read.delim("tx2gene.txt",stringsAsFactors = F)
简单两分组的比较
### 定义差异比较函数
# condition_ref为对照组, condition_treat为处理组
# level: 'transcript' 或者 'gene'
# bootstrap_summary默认不使用,QTL,plot_bootstrap需要
diff_exp_sleuth <- function(condition_ref, condition_treat, level, bootstrap_summary = FALSE) {
tab_metadata <- s2c
rownames(tab_metadata) <- tab_metadata$sample
conditions <- c(condition_ref, condition_treat)
# 选取待处理的两组样品
tab_metadata <- tab_metadata[tab_metadata$condition %in% conditions, ]
cond <- factor(tab_metadata$condition, levels = conditions) # ordered = T
md <- model.matrix(~cond,tab_metadata)
# sleuth_prep
if (level == 'gene') {
so <- sleuth_prep(tab_metadata, md, target_mapping = tx2gene, aggregation_column = 'Symbol', extra_bootstrap_summary = bootstrap_summary)
} else if (level == 'transcript') {
so <- sleuth_prep(tab_metadata, md, target_mapping = tx2gene, extra_bootstrap_summary = bootstrap_summary)
} else {
stop(paste0(level," isn't a valid level!"))
}
# 使用condition设计矩阵回归
so <- sleuth_fit(so)
# 使用截距项回归
so <- sleuth_fit(so, ~1, 'reduced')
# likelihood ratio test(LRT)检验以上两个模型
# 找到受condition_treat影响转录本或者基因
so <- sleuth_lrt(so, 'reduced', 'full')
# Wald test(WT)提供变化倍数信息(自然对数形式)
# LRT统计功效比WT更强,所以结合LRT的p值等信息和
# WT的变化倍数信息
so <- sleuth_wt(so, paste0('cond',condition_treat))
# 输出标准化的转录本表达值(no needed for gene-level)
if (level == 'transcript') {
otab = paste0("normalized_abundance_", level, "_", condition_treat, "_", condition_ref, ".tsv")
write.table(kallisto_table(so), otab, sep = "\t", quote = FALSE, row.names = FALSE)
}
# 添加beta(b), beta's standard error(se_b)和mean expression in the samples(mean_obs)信息
# 输出合并后的变量,便于后续的筛选
res_lrt <- sleuth_results(so, 'reduced:full', test_type = 'lrt')
res_wt <- sleuth_results(so, paste0('cond',condition_treat))
res <<- merge(res_lrt, res_wt[, c('target_id', 'b', 'se_b', 'mean_obs')], on = 'target_id', sort = FALSE)
# 输出信息合并后的表格
otab = paste0("differential_expression_analysis_", level, "_", condition_treat, "_", condition_ref, ".tsv")
write.table(res, otab, sep = "\t", quote = FALSE, row.names = FALSE)
return(so)
}
执行差异分析
sleuth_object <- diff_exp_sleuth("whole cortex", "neuron", 'transcript',bootstrap_summary = T)
sleuth_object <- diff_exp_sleuth("whole cortex", "neuron", 'gene',bootstrap_summary = T)
plot_bootstrap(sleuth_object,"NM_001081178",color_by = 'condition')
### PCA分析
so <- sleuth_object
par(mfrow = c(1, 2))
p1 <- plot_pca(so, pc_x = 1L, pc_y = 2L, color_by = 'condition')
p2 <- plot_pc_variance(so, PC_relative = TRUE)
cowplot::plot_grid(p1, p2, align = "h")

Note if you are using an Ensembl transcriptome, the easiest way to create the tx2gene data.frame is to use the ensembldb packages. The annotation packages can be found by version number, and use the pattern EnsDb.Hsapiens.vXX. The transcripts function can be used with return.type="DataFrame", in order to obtain something like the df object constructed in the code chunk above. See the ensembldb package vignette for more details.
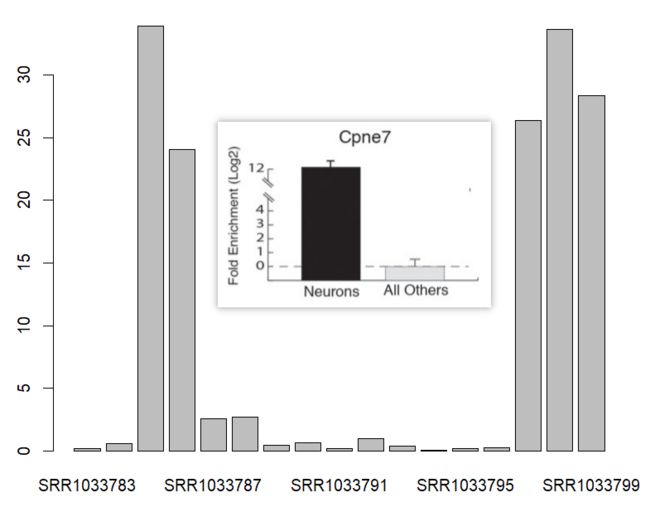
对比文献报道在不同组织类型验证的差异表达基因,发现sleuth的定量还是挺准确的。