背景介绍
随着癌症基因组学的进步,突变注释格式(MAF)被广泛接受并用于存储检测到的体细胞变体。 癌症基因组图谱项目对30多种不同的癌症进行了测序,每种癌症类型的样本量超过200种。由体细胞变体组成的结果数据以MAF格式形式存储。 只要数据采用MAF格式,该软件包就会尝试从TCGA源或任何内部研究中有效地汇总,分析,注释和可视化MAF文件.
准备
使用前要先将文件转换为maf格式,对于VCF格式文件,可以使用vcf2maf进行格式转换.
maf文件包含的内容:
Mandatory fields(必填项): Hugo_Symbol, Chromosome, Start_Position, End_Position, Reference_Allele, Tumor_Seq_Allele2, Variant_Classification, Variant_Type and Tumor_Sample_Barcode.
Recommended optional fields(选填项): non MAF specific fields containing VAF (Variant Allele Frequecy) and amino acid change information.
格式转换
#将突变结果进行注释,得到txt文件
for i in *.somatic.anno;do perl ~/software/Desktop/annovar/table_annovar.pl $sra_file /home/yang.zou/database/humandb_new/ -buildver hg19 -out variants --otherinfo -remove -protocol ensGene -operation g -nastring NA -outfile;done
#然后将所有.hg19_multianno.txt文件添加一列填入文件名前缀并将所有txt文件拼接成一个文件,提取出含有外显子的信息
for i in *.hg19_multianno.txt;do sed '1d' $i | sed "s/$/${i%%.*}/" >> all_annovar;done
grep -P "\texonic\t" all_annovar > all_annovar2
#格式转换
perl to-maftools.pl all_annovar2 #将文件转换为maf格式
#to-maftools.pl
use strict;
use warnings;
open (FA,"all_annovar2");
open (FB,">all_annovar3");
print FB "Chr\tStart\tEnd\tRef\tAlt\tFunc.ensGene\tGene.ensGene\tGeneDetail.ensGene\tExonicFunc.ensGene\tAAChange.ensGene\tTumor_Sample_Barcode\n";
while (){
chomp;
my @l=split /\t/,$_;
print FB $l[0],"\t",$l[1],"\t",$l[2],"\t",$l[3],"\t",$l[4],"\t",$l[5],"\t",$l[6],"\t",$l[7],"\t",$l[8],"\t",$l[9],"\t",$l[10],"\n";
}
总体分析框架
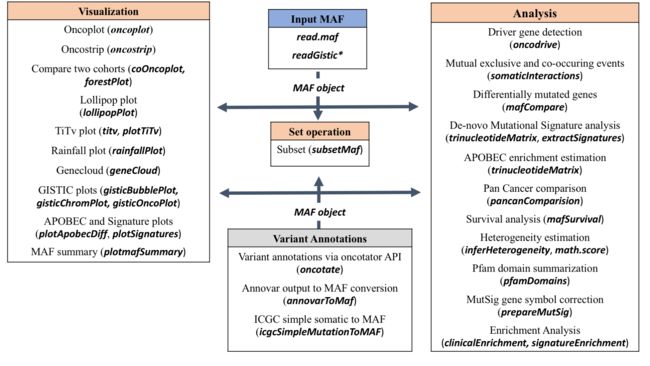
maftools安装
source("http://bioconductor.org/biocLite.R")
biocLite("maftools")
library(maftools)
注:安装过程特别麻烦,按了好几天,R版本要求3.3以上,也不要使用最新版本,可能有的包新版本还没同步。我使用的是:
version.string R version 3.4.1 (2017-06-30)
正式处理
读入annovar文件转换为maf——annovarToMaf
#read maf
var.annovar.maf = annovarToMaf(annovar = "all_annovar3", Center = 'NA', refBuild = 'hg19', tsbCol = 'Tumor_Sample_Barcode', table = 'ensGene',sep = "\t")
write.table(x=var.annovar.maf,file="var_annovar_maf",quote= F,sep="\t",row.names=F)
annovarToMaf函数说明
Description
Converts variant annotations from Annovar into a basic MAF.将annovar格式转换为maf格式
Usage
annovarToMaf(annovar, Center = NULL, refBuild = "hg19", tsbCol = NULL,
table = "refGene", basename = NULL, sep = "\t", MAFobj = FALSE,
sampleAnno = NULL)
Arguments
| 参数 |详细解释 |
| annovar| input annovar annotation file.|
| Center | Center field in MAF file will be filled with this value. Default NA.(MAF文件中的中心字段将填充此值。 默认NA)|
| refBuild | NCBI_Build field in MAF file will be filled with this value. Default hg19.(MAF文件中的NCBI_Build字段将填充此值。 默认hg19)|
| tsbCol | column name containing Tumor_Sample_Barcode or sample names in input file.(列名包含Tumor_Sample_Barcode或输入文件中的示例名称) |
| table | reference table used for gene-based annotations. Can be 'ensGene' or 'refGene'. Default 'refGene'(用于基于基因的注释的参考表。 可以是'ensGene'或'refGene'。 默认'refGene)|
| basename | If provided writes resulting MAF file to an output file. (将结果MAF文件写入输出文件)|
| sep | field seperator for input file. Default tab seperated.|
| MAFobj | If TRUE, returns results as an [MAF](http://127.0.0.1:37698/help/library/maftools/help/MAF object.|
| sampleAnno | annotations associated with each sample/Tumor_Sample_Barcode in input annovar file. If provided it will be included in MAF object. Could be a text file or a data.frame. Ideally annotation would contain clinical data, survival information and other necessary features associated with samples. Default NULL.(与输入annovar文件中的每个样本/ Tumor_Sample_Barcode相关联的注释。 如果提供,它将包含在MAF对象中。 可以是文本文件或data.frame。 理想情况下,注释将包含临床数据,生存信息和与样本相关的其他必要特征。 默认为NULL)|
然后用linux处理掉那些无义突变,也可以在后续设置参数去掉无义突变
sed 's/^NA/unknown/' var_annovar_maf > var_annovar_maf2
grep -v "^NA" var_annovar_maf | grep -v -P "\tUNKNOWN\t"> var_annovar_maf2
读入maf文件——read.maf
var_maf = read.maf(maf ="var_annovar_maf2")
plotmafSummary(maf = var_maf, rmOutlier = TRUE, addStat = 'median')
oncoplot(maf = var_maf, top = 400, writeMatrix=T,removeNonMutated = F,showTumorSampleBarcodes=T)
Description
Takes tab delimited MAF (can be plain text or gz compressed) file as an input and summarizes it in various ways. Also creates oncomatrix - helpful for visualization.
Usage
read.maf(maf, clinicalData = NULL, removeDuplicatedVariants = TRUE,
useAll = TRUE, gisticAllLesionsFile = NULL, gisticAmpGenesFile = NULL,
gisticDelGenesFile = NULL, gisticScoresFile = NULL, cnLevel = "all",
cnTable = NULL, isTCGA = FALSE, vc_nonSyn = NULL, verbose = TRUE)
Arguments
-
maf
tab delimited MAF file. File can also be gz compressed. Required. Alternatively, you can also provide already read MAF file as a dataframe.(制表符分隔的MAF文件。 文件也可以是gz压缩的。 也可以将已读取的MAF文件作为数据框提供
clinicalData
Clinical data associated with each sample/Tumor_Sample_Barcode in MAF. Could be a text file or a data.frame. Default NULL.(与MAF中每个样本/ Tumor_Sample_Barcode相关的临床数据。 可以是文本文件或data.frame。 默认为NULL)removeDuplicatedVariants
removes repeated variants in a particuar sample, mapped to multiple transcripts of same Gene. See Description. Default TRUE.(去除特定样本中的重复变体,映射到同一基因的多个转录本。 见说明。 默认值为TRUE)useAll
logical. Whether to use all variants irrespective of values in Mutation_Status. Defaults to TRUE. If FALSE, only uses with values Somatic.(逻辑。 是否使用所有变体而不管Mutation_Status中的值。 默认为TRUE。 如果为FALSE,则仅使用值Somatic)gisticAllLesionsFile
All Lesions file generated by gistic. e.g; all_lesions.conf_XX.txt, where XX is the confidence level. Default NULL.gisticAmpGenesFile
Amplification Genes file generated by gistic. e.g; amp_genes.conf_XX.txt, where XX is the confidence level. Default NULL.(扩增由gistic生成的基因文件。 例如; amp_genes.conf_XX.txt)gisticDelGenesFile
Deletion Genes file generated by gistic. e.g; del_genes.conf_XX.txt, where XX is the confidence level. Default NULL.(删除由gistic生成的Genes文件。 例如; del_genes.conf_XX.txt)gisticScoresFile
scores.gistic file generated by gistic. Default NULL(由gistic生成的scores.gistic文件)cnLevel
level of CN changes to use. Can be 'all', 'deep' or 'shallow'. Default uses all i.e, genes with both 'shallow' or 'deep' CN changes(CN级别改变使用。 可以是“全部”,“深层”或“浅层”。 默认使用所有,即具有“浅”或“深”CN变化的基因)cnTable
Custom copynumber data if gistic results are not available. Input file or a data.frame should contain three columns with gene name, Sample name and copy number status (either 'Amp' or 'Del'). Default NULL.(如果gistic结果不可用,则自定义copynumber数据。 输入文件或data.frame应包含三列,其中包含基因名称,样品名称和拷贝编号状态('Amp'或'Del')。 默认为NULL)isTCGA
Is input MAF file from TCGA source. If TRUE uses only first 12 characters from Tumor_Sample_Barcode.(是来自TCGA源的输入MAF文件。 如果TRUE仅使用Tumor_Sample_Barcode中的前12个字符。)vc_nonSyn
NULL. Provide manual list of variant classifications to be considered as non-synonymous. Rest will be considered as silent variants. Default uses Variant Classifications with High/Moderate variant consequences. http://asia.ensembl.org/Help/Glossary?id=535: "Frame_Shift_Del", "Frame_Shift_Ins", "Splice_Site", "Translation_Start_Site","Nonsense_Mutation", "Nonstop_Mutation", "In_Frame_Del","In_Frame_Ins", "Missense_Mutation"verbose
TRUE logical. Default to be talkative and prints summary.
绘制maf文件的摘要——plotmafSummary
该文件将每个样本中的变体数显示为堆积条形图,将变体类型显示为Variant_Classification汇总的箱形图。 我们可以在堆积的条形图中添加平均线或中线,以显示整个群组中变体的平均值/中值数
plotmafSummary(maf = laml, rmOutlier = TRUE, addStat = 'median', dashboard = TRUE, titvRaw = FALSE)
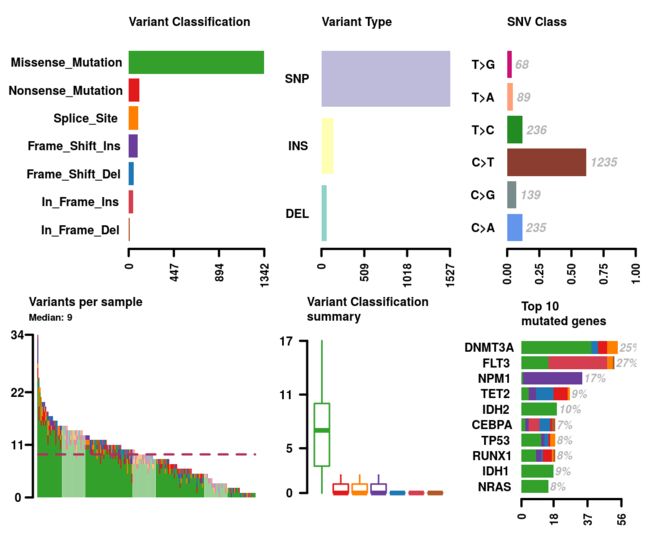
Description
Plots maf summary.
Usage
plotmafSummary(maf, file = NULL, rmOutlier = TRUE, dashboard = TRUE,
titvRaw = TRUE, width = 10, height = 7, addStat = NULL,
showBarcodes = FALSE, fs = 10, textSize = 2, color = NULL,
statFontSize = 3, titleSize = c(10, 8), titvColor = NULL, top = 10)
Arguments
maf
an MAF object generated by read.maffile
If given pdf file will be generated.rmOutlier
If TRUE removes outlier from boxplot.dashboard
If FALSE plots simple summary instead of dashboard style.titvRaw
TRUE. If false instead of raw counts, plots fraction.width
plot parameter for output file.height
plot parameter for output file.addStat
Can be either mean or median. Default NULL.showBarcodes
include sample names in the top bar plot.fs
base size for text. Default 10.color
named vector of colors for each Variant_Classification.titvColor
colors for SNV classifications.top
include top n genes dashboard plot. Default 10.
绘制瀑布图——oncoplots
Oncoplot函数使用“ComplexHeatmap”来绘制oncoplots2。 具体来说,oncoplot是ComplexHeatmap的OncoPrint功能的包装器,几乎没有任何修改和自动化,使绘图更容易。 侧面条形图和顶部条形图可分别由drawRowBar和drawColBar参数控制。
top的值需要视情况而定
oncoplot(maf = var_maf, top = 400, writeMatrix=T,removeNonMutated = F,showTumorSampleBarcodes=T)
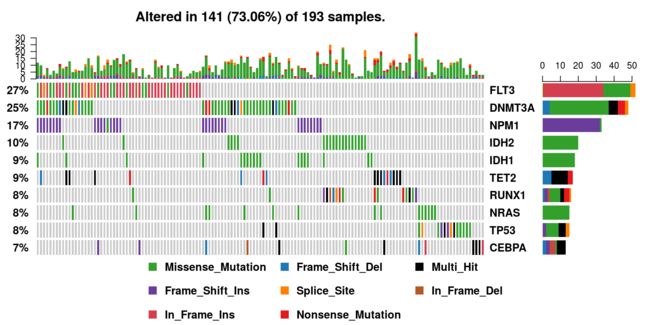
takes output generated by read.maf and draws an oncoplot
Usage
oncoplot(maf, top = 20, genes = NULL, mutsig = NULL, mutsigQval = 0.1,
drawRowBar = TRUE, drawColBar = TRUE, clinicalFeatures = NULL,
annotationDat = NULL, annotationColor = NULL, genesToIgnore = NULL,
showTumorSampleBarcodes = FALSE, removeNonMutated = TRUE, colors = NULL,
sortByMutation = FALSE, sortByAnnotation = FALSE,
annotationOrder = NULL, keepGeneOrder = FALSE, GeneOrderSort = TRUE,
sampleOrder = NULL, writeMatrix = FALSE, fontSize = 10,
SampleNamefontSize = 10, titleFontSize = 15, legendFontSize = 12,
annotationFontSize = 12, annotationTitleFontSize = 12)
Arguments
- maf
an MAF object generated by read.maf| - top
how many top genes to be drawn. defaults to 20.(显示多少基因)| - genes
Just draw oncoplot for these genes. Default NULL.(显示特定基因)| - mutsig
Mutsig resuts if availbale. Usually file named sig_genes.txt If provided plots significant genes and correpsonding Q-values as side row-bar. Default NULL.(如果可以的话,Mutsig会重新开始。 通常名为sig_genes.txt的文件如果提供,则绘制重要基因并将Q值作为侧行条对应。 默认为NULL) | - mutsigQval
Q-value to choose significant genes from mutsig results. Default 0.1(从mutsig结果中选择重要基因的Q值。 默认值为0.1)| - genesToIgnore
do not show these genes in Oncoplot. Default NULL. - showTumorSampleBarcodes
logical to include sample names.(逻辑包含样本名称) - removeNonMutated
Logical. IfTRUEremoves samples with no mutations in the oncoplot for better visualization. DefaultTRUE.(消除无义突变) - sortByMutation
Force sort matrix according mutations. Helpful in case of MAF was read along with copy number data. Default FALSE.(根据突变强制排序矩阵。 在阅读MAF的情况下有用以及拷贝数数据。 默认为FALSE)| - sortByAnnotation
logical sort oncomatrix (samples) by provided 'clinicalFeatures'. Sorts based on first 'clinicalFeatures'. Defaults to FALSE. column-sort| - annotationOrder
Manually specify order for annotations. Works only for first 'clinicalFeatures'. Default NULL. | - keepGeneOrder
logical whether to keep order of given genes. Default FALSE, order according to mutation frequency| - GeneOrderSort
logical this is applicable when 'keepGeneOrder' is TRUE. Default TRUE| - sampleOrder
Manually speify sample names for oncolplot ordering. Default NULL.| - writeMatrix
writes character coded matrix used to generate the plot to an output file. This can be used as an input for ComplexHeatmap oncoPrint function if you wish to customize the plot.(将用于生成绘图的字符编码矩阵写入输出文件。 如果您想自定义绘图,则可以将其用作ComplexHeatmap oncoPrint函数的输入)
通过包括与样本相关的注释(临床特征),改变变体分类的颜色并包括显着性的q值(从MutSig或类似程序生成),可以进一步改善Oncoplots。
col = RColorBrewer::brewer.pal(n = 8, name = 'Paired')
names(col) = c('Frame_Shift_Del','Missense_Mutation', 'Nonsense_Mutation', 'Multi_Hit', 'Frame_Shift_Ins','In_Frame_Ins', 'Splice_Site', 'In_Frame_Del')
#Color coding for FAB classification; try getAnnotations(x = laml) to see available annotations.
fabcolors = RColorBrewer::brewer.pal(n = 8,name = 'Spectral')
names(fabcolors) = c("M0", "M1", "M2", "M3", "M4", "M5", "M6", "M7")
fabcolors = list(FAB_classification = fabcolors)
#MutSig reusults
laml.mutsig <- system.file("extdata", "LAML_sig_genes.txt.gz", package = "maftools")
oncoplot(maf = laml, colors = col, mutsig = laml.mutsig, mutsigQval = 0.01, clinicalFeatures = 'FAB_classification', sortByAnnotation = TRUE, annotationColor = fabcolors)
[图片上传失败...(image-fc6334-1536734778754)]
使用oncostrip函数可视化任何一组基因,它们在每个样本中绘制类似于cBioPortal上的OncoPrinter工具的突变。 oncostrip可用于使用top或gene参数绘制任意数量的基因
#显示特定基因
oncostrip(maf = laml, genes = c('DNMT3A','NPM1', 'RUNX1'))

绘制箱线图—— titv
titv函数将SNP分类为Transitions_vs_Transversions,并以各种方式返回汇总表的列表。 汇总数据也可以显示为一个箱线图,显示六种不同转换的总体分布,并作为堆积条形图显示每个样本中的转换比例。

Description
takes output generated by read.maf and classifies Single Nucleotide Variants into Transitions and Transversions.
Usage
titv(maf, useSyn = FALSE, plot = TRUE, file = NULL)
Arguments
maf
an MAF object generated by read.mafuseSyn
Logical. Whether to include synonymous variants in analysis. Defaults to FALSE.
-
plot
plots a titv fractions. default TRUE.
-
file
basename for output file name. If given writes summaries to output file. Default NULL.
#绘制棒棒图——lollipopPlot
棒棒糖图是简单且最有效的方式,显示蛋白质结构上的突变点。许多致癌基因具有比任何其他基因座更频繁突变的优先位点。这些斑点被认为是突变热点,棒棒糖图可以用于显示它们以及其他突变。我们可以使用函数lollipopPlot绘制这样的图。这个功能要求我们在maf文件中有氨基酸改变信息。然而,MAF文件没有关于命名氨基酸变化字段的明确指南,不同的研究具有不同的氨基酸变化的字段(或列)名称。默认情况下,lollipopPlot查找列AAChange,如果在MAF文件中找不到它,则会打印所有可用字段并显示警告消息。对于以下示例,MAF文件包含字段/列名称“Protein_Change”下的氨基酸变化。我们将使用参数AACol手动指定它。此函数还将绘图作为ggplot对象返回,如果需要,用户稍后可以修改该对象。
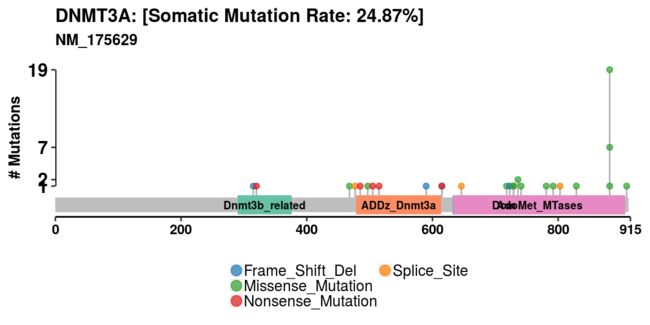
maftools还可以制作很多图,比如
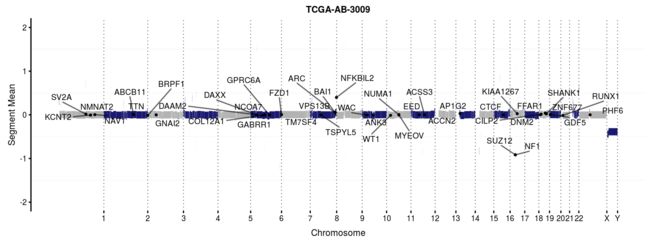
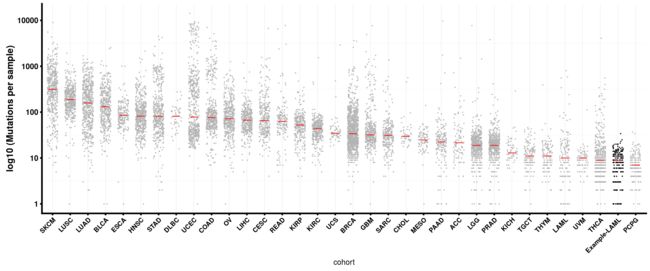
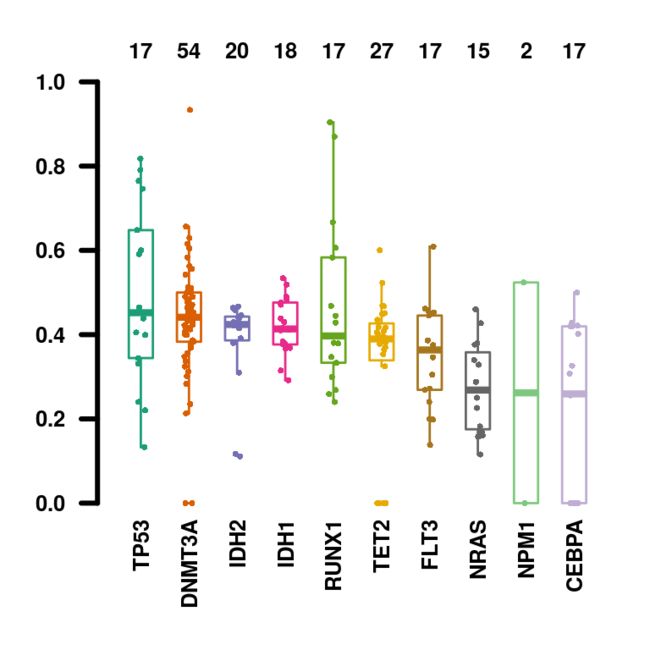
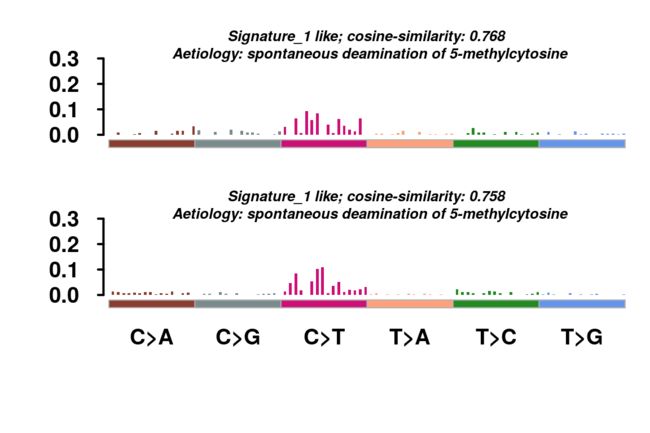
还可以用函数geneCloud绘制突变基因的词云图。 每个基因的大小与其突变/改变的样品总数成比例。

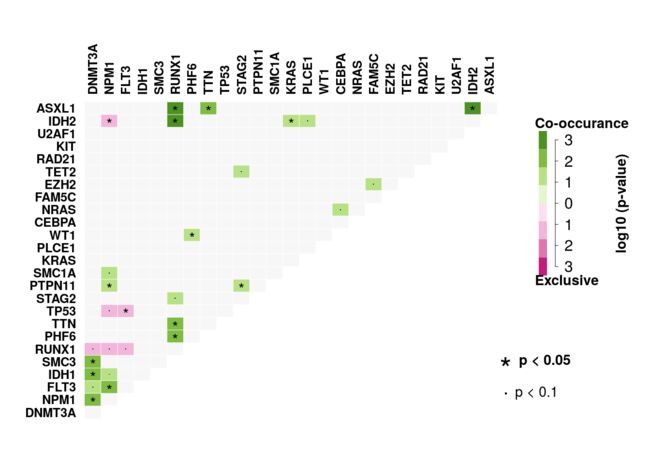
癌症中的许多引起疾病的基因共同发生或在其突变模式中显示出强烈的排他性。 可以使用somaticInteractions函数检测这种相互排斥或共同发生的基因组,其执行成对的Fisher's Exact检验以检测这种显着的基因对。 somaticInteractions函数还使用cometExactTest来识别涉及> 2个基因的潜在改变的基因集
maftools包 功能很强大,具体可参考:
http://bioconductor.org/packages/release/bioc/vignettes/maftools/inst/doc/maftools.html