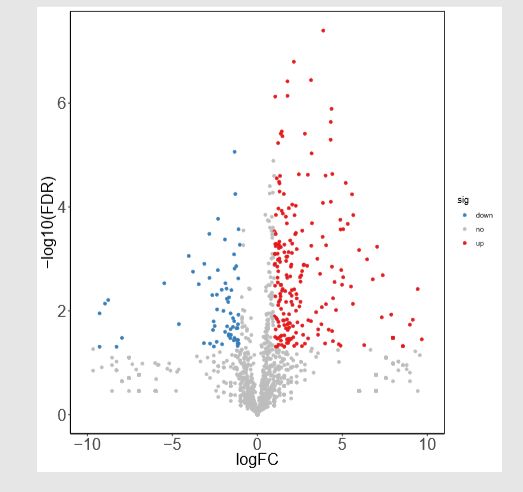
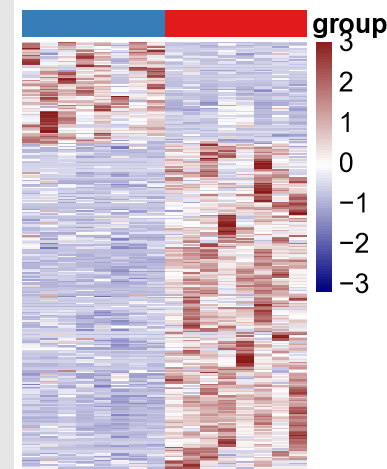
- C语言数据结构与算法专栏目录
CodeAllen嵌入式
嵌入式C语言数据结构算法
后序会开一个《嵌入式数据结构专栏》主要为了学习嵌入式的同学,软件能力提升和大厂面试能力,感谢大家关注!直达专栏:https://blog.csdn.net/super828/category_11083370.html《C语言数据结构与算法》专栏已经更新完毕,共计72篇分享,后期会逐渐修改错误并添加内容0数据之间的关系有哪些?1如何度量一个算法的好坏?2常见的时间复杂度实例
- 2025.7.4总结
天真小巫
职场记录职场和发展
感恩环节:感谢今日工作顺利度过,明天终于能美美的睡个懒觉了。感谢这周有个美好的双休。今日去实验室参观设备,感谢我的一个同事解答了我关于硬件设备与所做软件业务之间的关系,通过控制器控制网元等相关设备,同时,虽然参加过两周的硬装培训,但在这个光交箱得众多设备里,连交换机长什么样子都忘了。同事之间的交流完全插不上话。业务上还是需要多学习。如果所学的只是不能为自己所用,那么它将化为一摊死水。有氧运动:晚上
- 洛谷分支结构题单刷题记录 java
阿乌阿呜
JAVA刷题记录java算法
目录P2433【深基1-2】小学数学N合一P5709【深基2.习6】ApplesPrologue/苹果和虫子P5710【深基3.例2】数的性质P5711【深基3.例3】闰年判断P5712【深基3.例4】ApplesP5713【深基3.例5】洛谷团队系统P5714【深基3.例7】肥胖问题P5715【深基3.例8】三位数排序P5716【深基3.例9】月份天数P1085[NOIP2004普及组]不高兴的
- Leetcode刷题java之520(检测大写字母)
qq_42342642
leetcodejava算法
执行结果:通过显示详情添加备注执行用时:1ms,在所有Java提交中击败了98.19%的用户内存消耗:36.8MB,在所有Java提交中击败了57.52%的用户通过测试用例:550/550
- MySQL分区
我说人人平等
mysqlmysql分区
MySQL分区优点:1,和单个磁盘或者文件系统分区相比,可以存储更多数据2,优化查询。在where子句中包含分区条件时,可以只扫描必要的一个或者多个分区来提高查询效率;同时涉及sum()和count()这类聚合查询时,可以容易的在每个分区上并行处理,最终只需要汇总所有分区得到的结果3,对于已经过期或者不需要保存的数据,可以通过删除与这些数据有关的分区来快速删除数据4,跨多个磁盘来分散数据查询,以获
- JDBC连接池
今惜时
JDBC数据库javamysql
数据库连接池什么是连接池连接池是创建和管理一个连接的缓冲池的技术,这些连接准备好被任何需要它们的线程使用。这种连接“汇集”起来的技术基于这样的一个事实:对于大多数应用程序,当它们正在处理通常需要数毫秒完成的事务时,仅需要能够访问JDBC连接的1个线程。当不处理事务时,这个连接就会闲置。相反,连接池允许闲置的连接被其它需要的线程使用。事实上,当一个线程需要用JDBC对一个GBase或其它数据库操作时
- 21天刷题计划之10.1—统计大写字母个数(Java语言描述)
justlikeu777
21天刷题计划java基础算法基础
题目描述:找出给定字符串中大写字符(即’A’-‘Z’)的个数接口说明原型:intCalcCapital(Stringstr);返回值:int输入描述:输入一个String数据输出描述:输出string中大写字母的个数示例1输入add123#$%#%#O输出1分析:获取输出的字符串,将字符串转换成字符数组,遍历字符数组并判断是否为大写字母即可。importjava.util.Scanner;publ
- 【云原生篇】微服务革命:解锁Istio与Service Mesh
林木森^~^
云原生云原生微服务istio
ServiceMeshServiceMesh是一种用于处理服务间通信的基础设施层,它以轻量级的网络代理的形式实现,这些代理与应用程序的微服务一同部署。ServiceMesh的核心目的是将网络通信的复杂性从应用程序代码中抽象出来,从而使开发人员可以专注于业务逻辑的开发,而不是通信的细节和问题。主要特点和功能服务发现:自动管理服务间的发现,使得各服务可以相互识别并进行通信。负载均衡:智能地将请求流量分
- 分布式ID设计方案详解:从理论到实践
一、为什么需要分布式ID?在分布式系统中,唯一ID的生成面临两大核心挑战:全局唯一性:避免跨节点、跨数据中心的ID冲突。有序性:确保ID按时间或业务规则递增,提升数据库写入性能(如InnoDB的B+树索引)。传统单机自增ID(如MySQLAUTO_INCREMENT)无法满足分库分表、高并发等场景需求,因此需引入分布式ID方案。二、主流分布式ID方案对比方案优点缺点适用场景UUID简单、无中心化依
- 也谈一下 30+ 程序员的出路
写编程的木木
langchain产品经理python开发语言大模型
前言前两天和一个前端同学聊天,他说不准备再做前端了,准备去考公。不过难度也很大。从20152016年那会儿开始互联网行业爆发,到现在有7、8年了,当年20多岁的小伙子们,现在也都30+了大量的人面临这个问题:大龄程序员就业竞争力差,未来该如何安身立命?先说我个人的看法:除非你有其他更好的资源,否则没有更好的出路认真搞技术,保持技术能力,你大概率不会失业(至少外包还在招人,外包也不少挣…)考公之我见
- 介绍6款密码暴力破解工具
网安导师小李
程序员网络安全编程web安全网络安全tcp/ipphppythonjava
暴力破解就是通过不断穷举可能的密码,直至密码验证成功,暴力破解分为密码爆破和密码喷洒,密码爆破就是不断的去尝试不同的密码,密码喷洒就是通过已知密码不断去尝试账号。下面介绍6款常见的暴力破解工具。01hydraHydra(九头蛇)是THC组织开发的,是一款非常流行的密码破解工具,可以对多种服务的账号和密码进行爆破,包括Web登录、数据库、SSH、FTP等服务,支持Linux、Windows、Mac平
- 下一代防火墙
999感冒灵.
网络安全
一.防火墙是什么1.防火墙的定义:防火墙是一个位于内部网络与外部网络之间的安全系统(网络中不同区域之间),是按照一定的安全策略建立起来的硬件或软件系统,用于流量控制的系统(隔离),保护内部网络资源免受威胁(保护)。防火墙的主要用于防止黑客对安全区域网络的攻击,保护内部网络的安全运行。2.防火墙基本性质:①安全区域和接口:一台防火墙具有多个接口每个接口属于一个安全区域,每个区域具有唯一的名称,所以防
- 发起请求并处理响应:`XHR` 与 `axios` 使用指南来啦[特殊字符]~
又又又要长脑子呐~了解到通过发起HTTP请求并在不刷新页面的情况下更新页面内容是一种常见的需求。学习使用XMLHttpRequest或axios来实现,现在进行对比两者,比较项目使用时候的优缺点,文末使用表格进行对比学习1.使用XHR实现下面是一个使用XMLHttpRequest发起GET请求并处理服务器响应的示例:html体验AI代码助手代码解读复制代码//创建一个新的XMLHttpReques
- 2024年最全kali无线渗透之用wps加密模式可破解wpa模式的密码12_kali wps,网络安全开发究竟该如何学习
2401_84558314
程序员wpsweb安全学习
一、网安学习成长路线图网安所有方向的技术点做的整理,形成各个领域的知识点汇总,它的用处就在于,你可以按照上面的知识点去找对应的学习资源,保证自己学得较为全面。二、网安视频合集观看零基础学习视频,看视频学习是最快捷也是最有效果的方式,跟着视频中老师的思路,从基础到深入,还是很容易入门的。三、精品网安学习书籍当我学到一定基础,有自己的理解能力的时候,会去阅读一些前辈整理的书籍或者手写的笔记资料,这些笔
- 计算机网络(网页显示过程,TCP三次握手,HTTP1.0,1.1,2.0,3.0,JWT cookie)
老虎0627
计算机网络计算机网络tcp/ip网络协议
前言最近一直在看后端开发的面经,里面涉及到了好多计算机网络的知识,在这里以问题的形式写一个学习笔记(其中参考了:JavaGuide和小林coding这两个很好的学习网站)1.当键入网址后,到网页显示,其间发生了什么?(1)首先浏览器会解析URL。(如确定协议像Http或Https)(2)然后通过DNS服务器把域名解析为IP地址。(找到服务器啦)(3)接着TCP协议三次握手和服务器建立连接。(客户端
- MySQL分布式ID冲突详解:场景、原因与解决方案
码不停蹄的玄黓
mysql分布式数据库ID冲突
引言在分布式系统开发中,你是否遇到过这样的崩溃时刻?——明明每个数据库实例的自增ID都从1开始,插入数据时却提示“Duplicateentry‘100’forkey‘PRIMARY’”;或者分库分表后,不同库里的订单ID竟然重复,业务合并时直接报错……这些问题的核心,都是分布式ID冲突。今天咱们就来扒一扒MySQL分布式ID冲突的常见场景、底层原因,以及对应的解决方案,帮你彻底避开这些坑!一、为什
- MongoDB Rust驱动代码架构深度解析
倪俪珍Phineas
MongoDBRust驱动代码架构深度解析mongo-rust-driverTheofficialMongoDBRustDriver项目地址:https://gitcode.com/gh_mirrors/mo/mongo-rust-driver前言本文将对MongoDB官方Rust驱动(mongo-rust-driver)的核心架构进行深入解析,帮助开发者理解其设计哲学和实现细节。我们将从客户端构
- 如何在YashanDB中管理数据模型变更
数据库
在现代企业中,数据模型的变更管理扮演着关键角色。无论是扩展现有业务,还是应对新的需求,业务模型的改变往往需要相应的数据模型更新。如何有效地管理这些变更,确保数据的完整性、一致性及应用的高可用性,成为了数据架构师和开发者必须面对的重要问题。本文将详细探讨在YashanDB中管理数据模型变更的策略和方法,旨在提升对YashanDB数据库技术的理解及应用能力。数据模型变更管理的关键要素版本控制与变更日志
- 如何在YashanDB数据库中使用JSON数据类型?
数据库
随着海量结构化与半结构化数据的快速增长,关系型数据库面临性能瓶颈和数据一致性的挑战。JSON作为一种灵活的半结构化数据格式,在多领域数据交换和存储中广泛应用。YashanDB作为支持多种存储结构和高性能事务处理的数据库产品,提供了对JSON数据类型的支持,以满足现代复杂业务对半结构化数据处理的需求。本文旨在基于YashanDB体系架构及存储引擎特性,深入解析JSON数据类型的技术原理与实现方式,为
- 如何在YashanDB数据库中实现数据查询优化
数据库
在现代信息技术环境中,数据量的快速增长使得数据库的性能优化成为重要课题。如何提升查询速度,降低资源消耗,成为了数据库管理人员和开发者必须面对的挑战。有效的数据查询优化不仅能提高响应时间,还能显著提升用户体验与系统效率。在YashanDB数据库中,优化数据查询需从多个技术角度进行综合考量与实际应用。利用索引技术优化查询索引是提升数据库查询性能的常用手段。在YashanDB中,主要支持BTree索引、
- 如何在YashanDB数据库中实现数据模型的简化
数据库
在现代数据库技术领域,数据模型的复杂性经常导致性能瓶颈和维护困惑。随着数据规模的增长和业务诉求的增加,复杂的数据结构、冗余的存储和不必要的关联关系都会影响整体数据库的性能和可维护性。特别是在面对动态变化的业务需求时,灵活性和扩展性成为关键因素。YashanDB提供了一系列功能强大的工具和机制,能够有效简化数据模型,提升数据库性能,并增强数据操作的灵活性。本文章旨在为数据库开发者和架构师提供技术洞见
- 如何在YashanDB数据库中实现复杂事务管理
数据库
在现代数据库管理系统中,事务管理是一项关键功能。复杂的事务管理可以确保多条SQL操作的原子性、一致性、隔离性和持久性(ACID特性),减少数据的不一致和错误。尤其在高并发场景中,事务管理的机制与实现至关重要。因此,构建高效的事务管理系统,对于提升数据库的性能及应用程序的可靠性具有深远影响。YashanDB的事务特性YashanDB数据库支持全面的事务管理功能,通过多版本并发控制(MVCC)、事务隔
- 如何在YashanDB数据库中管理用户权限
数据库
在数据库管理系统中,用户权限的管理是保障数据安全和系统稳定运行的关键环节。合理的权限控制能有效防止未经授权的访问和误操作,同时满足业务需求的灵活性。对于YashanDB数据库,充分理解其权限体系与管理机制,有助于构建安全、稳定且高效的数据库应用环境。本文将深入解析YashanDB中用户权限管理的技术原理、实现功能和最佳实践。YashanDB的用户与角色机制YashanDB管理权限的核心实体为“用户
- 如何在YashanDB数据库中进行高效的JSON数据存储
数据库
随着业务对非结构化和半结构化数据存储需求的增加,JSON数据类型逐渐成为数据库支持的关键特性。然而,JSON数据的高效存储与访问面临性能瓶颈、一致性保障及空间利用率等挑战。YashanDB作为现代企业级数据库,需提供有效的机制解决上述难题,从而满足实时查询、高并发访问及数据一致性的需求。本文针对YashanDB数据库的体系架构、存储引擎及索引机制,深入分析如何实现高效的JSON数据存储与访问,旨在
- 如何在YashanDB数据库中高效处理海量数据
数据库
在现代数据库技术中,海量数据的管理和处理成为了一个普遍存在的挑战。随着数据规模的不断扩大,性能瓶颈、数据一致性问题以及易用性需求等问题日益凸显。这些挑战促使企业寻求更为高效的解决方案,以支撑海量数据的存储、分析与挖掘。YashanDB作为一款专为处理海量数据而设计的数据库,凭借其高可扩展性、高并发性能和高可用性,提供了一系列技术手段以应对这些挑战。本文旨在探讨如何在YashanDB中高效地管理和处
- 如何有效管理YashanDB的访问控制
数据库
引言在当今数字化的业务环境中,数据安全性和访问控制是数据库管理的核心问题。随着数据规模的不断扩大,以及对数据隐私和合规性的要求日益增强,如何有效管理数据库的访问权限已成为企业面临的重大挑战。YashanDB作为一个高性能的数据库管理系统,具备丰富的访问控制功能,但同时也带来了复杂的管理需求。本篇文章将深入探讨YashanDB的访问控制机制,包括用户管理、角色权限、身份认证及其他相关策略,旨在为数据
- 如何在YashanDB数据库中保持数据一致性与完整性
数据库
在现代数据库管理系统中,确保数据的一致性与完整性是面临的主要挑战之一。这一挑战在高并发、高要求的数据操作场景中尤为突出。YashanDB作为一种高性能的分布式数据库,采用了多种技术手段以保持数据的一致性与完整性。本文将深入探讨YashanDB中实现数据一致性与完整性的核心技术原理,适用于对高并发和复杂事务有一定理解的数据库管理员(DBA)和开发人员。事务管理与ACID特性事务是数据库操作的基本单元
- 如何实现YashanDB中的数据冗余处理
数据库
数据冗余是数据库管理中的一个重要话题,直接影响到数据的可用性与可靠性。在高并发场景下,数据冗余能够有效防止数据丢失,并提升系统的容灾能力。YashanDB作为一款高性能的数据库产品,通过灵活的结构和多种部署方式,实现了数据冗余处理。本文将详细探讨YashanDB中实现数据冗余处理的技术细节,为数据库管理员和开发人员提供理论支持和实践指导。YashanDB的数据冗余机制单机部署中的数据冗余在单机部署
- 如何确保YashanDB数据库的性能稳定?
数据库
在当今数据量激增的背景下,数据库的性能稳定性成为企业技术架构成功的关键因素之一。数据库面临的挑战包括性能瓶颈、数据一致性问题及并发访问的影响。为了应对这些问题,YashanDB作为一种新兴的数据库管理系统,提供了先进的架构和功能,旨在为高性能和高可用性提供保障。本文将详细探讨确保YashanDB数据库性能稳定性的方法,旨在为数据库管理员、系统架构师及IT技术负责人提供实用建议,实现企业数据处理的高
- 如何设计基于YashanDB数据库的高效查询
数据库
在当今数据驱动的业务环境中,提高数据库查询性能已经成为各类企业面临的重大挑战。随着数据量的快速增长,许多机构遭遇了性能瓶颈、数据一致性问题和查询响应延迟等一系列问题。在这样的背景下,优化数据库架构、提高查询效率迫在眉睫。本文将集中在YashanDB数据库的查询设计上,提供技术分析和操作指导,以帮助开发人员设计高效的查询策略,实现优越的性能。YashanDB的体系架构YashanDB支持多种部署形态
- sql统计相同项个数并按名次显示
朱辉辉33
javaoracle
现在有如下这样一个表:
A表
ID Name time
------------------------------
0001 aaa 2006-11-18
0002 ccc 2006-11-18
0003 eee 2006-11-18
0004 aaa 2006-11-18
0005 eee 2006-11-18
0004 aaa 2006-11-18
0002 ccc 20
- Android+Jquery Mobile学习系列-目录
白糖_
JQuery Mobile
最近在研究学习基于Android的移动应用开发,准备给家里人做一个应用程序用用。向公司手机移动团队咨询了下,觉得使用Android的WebView上手最快,因为WebView等于是一个内置浏览器,可以基于html页面开发,不用去学习Android自带的七七八八的控件。然后加上Jquery mobile的样式渲染和事件等,就能非常方便的做动态应用了。
从现在起,往后一段时间,我打算
- 如何给线程池命名
daysinsun
线程池
在系统运行后,在线程快照里总是看到线程池的名字为pool-xx,这样导致很不好定位,怎么给线程池一个有意义的名字呢。参照ThreadPoolExecutor类的ThreadFactory,自己实现ThreadFactory接口,重写newThread方法即可。参考代码如下:
public class Named
- IE 中"HTML Parsing Error:Unable to modify the parent container element before the
周凡杨
html解析errorreadyState
错误: IE 中"HTML Parsing Error:Unable to modify the parent container element before the child element is closed"
现象: 同事之间几个IE 测试情况下,有的报这个错,有的不报。经查询资料后,可归纳以下原因。
- java上传
g21121
java
我们在做web项目中通常会遇到上传文件的情况,用struts等框架的会直接用的自带的标签和组件,今天说的是利用servlet来完成上传。
我们这里利用到commons-fileupload组件,相关jar包可以取apache官网下载:http://commons.apache.org/
下面是servlet的代码:
//定义一个磁盘文件工厂
DiskFileItemFactory fact
- SpringMVC配置学习
510888780
springmvc
spring MVC配置详解
现在主流的Web MVC框架除了Struts这个主力 外,其次就是Spring MVC了,因此这也是作为一名程序员需要掌握的主流框架,框架选择多了,应对多变的需求和业务时,可实行的方案自然就多了。不过要想灵活运用Spring MVC来应对大多数的Web开发,就必须要掌握它的配置及原理。
一、Spring MVC环境搭建:(Spring 2.5.6 + Hi
- spring mvc-jfreeChart 柱图(1)
布衣凌宇
jfreechart
第一步:下载jfreeChart包,注意是jfreeChart文件lib目录下的,jcommon-1.0.23.jar和jfreechart-1.0.19.jar两个包即可;
第二步:配置web.xml;
web.xml代码如下
<servlet>
<servlet-name>jfreechart</servlet-nam
- 我的spring学习笔记13-容器扩展点之PropertyPlaceholderConfigurer
aijuans
Spring3
PropertyPlaceholderConfigurer是个bean工厂后置处理器的实现,也就是BeanFactoryPostProcessor接口的一个实现。关于BeanFactoryPostProcessor和BeanPostProcessor类似。我会在其他地方介绍。PropertyPlaceholderConfigurer可以将上下文(配置文件)中的属性值放在另一个单独的标准java P
- java 线程池使用 Runnable&Callable&Future
antlove
javathreadRunnablecallablefuture
1. 创建线程池
ExecutorService executorService = Executors.newCachedThreadPool();
2. 执行一次线程,调用Runnable接口实现
Future<?> future = executorService.submit(new DefaultRunnable());
System.out.prin
- XML语法元素结构的总结
百合不是茶
xml树结构
1.XML介绍1969年 gml (主要目的是要在不同的机器进行通信的数据规范)1985年 sgml standard generralized markup language1993年 html(www网)1998年 xml extensible markup language
- 改变eclipse编码格式
bijian1013
eclipse编码格式
1.改变整个工作空间的编码格式
改变整个工作空间的编码格式,这样以后新建的文件也是新设置的编码格式。
Eclipse->window->preferences->General->workspace-
- javascript中return的设计缺陷
bijian1013
JavaScriptAngularJS
代码1:
<script>
var gisService = (function(window)
{
return
{
name:function ()
{
alert(1);
}
};
})(this);
gisService.name();
&l
- 【持久化框架MyBatis3八】Spring集成MyBatis3
bit1129
Mybatis3
pom.xml配置
Maven的pom中主要包括:
MyBatis
MyBatis-Spring
Spring
MySQL-Connector-Java
Druid
applicationContext.xml配置
<?xml version="1.0" encoding="UTF-8"?>
&
- java web项目启动时自动加载自定义properties文件
bitray
javaWeb监听器相对路径
创建一个类
public class ContextInitListener implements ServletContextListener
使得该类成为一个监听器。用于监听整个容器生命周期的,主要是初始化和销毁的。
类创建后要在web.xml配置文件中增加一个简单的监听器配置,即刚才我们定义的类。
<listener>
<des
- 用nginx区分文件大小做出不同响应
ronin47
昨晚和前21v的同事聊天,说到我离职后一些技术上的更新。其中有个给某大客户(游戏下载类)的特殊需求设计,因为文件大小差距很大——估计是大版本和补丁的区别——又走的是同一个域名,而squid在响应比较大的文件时,尤其是初次下载的时候,性能比较差,所以拆成两组服务器,squid服务于较小的文件,通过pull方式从peer层获取,nginx服务于较大的文件,通过push方式由peer层分发同步。外部发布
- java-67-扑克牌的顺子.从扑克牌中随机抽5张牌,判断是不是一个顺子,即这5张牌是不是连续的.2-10为数字本身,A为1,J为11,Q为12,K为13,而大
bylijinnan
java
package com.ljn.base;
import java.util.Arrays;
import java.util.Random;
public class ContinuousPoker {
/**
* Q67 扑克牌的顺子 从扑克牌中随机抽5张牌,判断是不是一个顺子,即这5张牌是不是连续的。
* 2-10为数字本身,A为1,J为1
- 翟鸿燊老师语录
ccii
翟鸿燊
一、国学应用智慧TAT之亮剑精神A
1. 角色就是人格
就像你一回家的时候,你一进屋里面,你已经是儿子,是姑娘啦,给老爸老妈倒怀水吧,你还觉得你是老总呢?还拿派呢?就像今天一样,你们往这儿一坐,你们之间是什么,同学,是朋友。
还有下属最忌讳的就是领导向他询问情况的时候,什么我不知道,我不清楚,该你知道的你凭什么不知道
- [光速与宇宙]进行光速飞行的一些问题
comsci
问题
在人类整体进入宇宙时代,即将开展深空宇宙探索之前,我有几个猜想想告诉大家
仅仅是猜想。。。未经官方证实
1:要在宇宙中进行光速飞行,必须首先获得宇宙中的航行通行证,而这个航行通行证并不是我们平常认为的那种带钢印的证书,是什么呢? 下面我来告诉
- oracle undo解析
cwqcwqmax9
oracle
oracle undo解析2012-09-24 09:02:01 我来说两句 作者:虫师收藏 我要投稿
Undo是干嘛用的? &nb
- java中各种集合的详细介绍
dashuaifu
java集合
一,java中各种集合的关系图 Collection 接口的接口 对象的集合 ├ List 子接口 &n
- 卸载windows服务的方法
dcj3sjt126com
windowsservice
卸载Windows服务的方法
在Windows中,有一类程序称为服务,在操作系统内核加载完成后就开始加载。这里程序往往运行在操作系统的底层,因此资源占用比较大、执行效率比较高,比较有代表性的就是杀毒软件。但是一旦因为特殊原因不能正确卸载这些程序了,其加载在Windows内的服务就不容易删除了。即便是删除注册表中的相 应项目,虽然不启动了,但是系统中仍然存在此项服务,只是没有加载而已。如果安装其他
- Warning: The Copy Bundle Resources build phase contains this target's Info.plist
dcj3sjt126com
iosxcode
http://developer.apple.com/iphone/library/qa/qa2009/qa1649.html
Excerpt:
You are getting this warning because you probably added your Info.plist file to your Copy Bundle
- 2014之C++学习笔记(一)
Etwo
C++EtwoEtwoiterator迭代器
已经有很长一段时间没有写博客了,可能大家已经淡忘了Etwo这个人的存在,这一年多以来,本人从事了AS的相关开发工作,但最近一段时间,AS在天朝的没落,相信有很多码农也都清楚,现在的页游基本上达到饱和,手机上的游戏基本被unity3D与cocos占据,AS基本没有容身之处。so。。。最近我并不打算直接转型
- js跨越获取数据问题记录
haifengwuch
jsonpjsonAjax
js的跨越问题,普通的ajax无法获取服务器返回的值。
第一种解决方案,通过getson,后台配合方式,实现。
Java后台代码:
protected void doPost(HttpServletRequest req, HttpServletResponse resp)
throws ServletException, IOException {
String ca
- 蓝色jQuery导航条
ini
JavaScripthtmljqueryWebhtml5
效果体验:http://keleyi.com/keleyi/phtml/jqtexiao/39.htmHTML文件代码:
<!DOCTYPE html>
<html xmlns="http://www.w3.org/1999/xhtml">
<head>
<title>jQuery鼠标悬停上下滑动导航条 - 柯乐义<
- linux部署jdk,tomcat,mysql
kerryg
jdktomcatlinuxmysql
1、安装java环境jdk:
一般系统都会默认自带的JDK,但是不太好用,都会卸载了,然后重新安装。
1.1)、卸载:
(rpm -qa :查询已经安装哪些软件包;
rmp -q 软件包:查询指定包是否已
- DOMContentLoaded VS onload VS onreadystatechange
mutongwu
jqueryjs
1. DOMContentLoaded 在页面html、script、style加载完毕即可触发,无需等待所有资源(image/iframe)加载完毕。(IE9+)
2. onload是最早支持的事件,要求所有资源加载完毕触发。
3. onreadystatechange 开始在IE引入,后来其它浏览器也有一定的实现。涉及以下 document , applet, embed, fra
- sql批量插入数据
qifeifei
批量插入
hi,
自己在做工程的时候,遇到批量插入数据的数据修复场景。我的思路是在插入前准备一个临时表,临时表的整理就看当时的选择条件了,临时表就是要插入的数据集,最后再批量插入到数据库中。
WITH tempT AS (
SELECT
item_id AS combo_id,
item_id,
now() AS create_date
FROM
a
- log4j打印日志文件 如何实现相对路径到 项目工程下
thinkfreer
Weblog4j应用服务器日志
最近为了实现统计一个网站的访问量,记录用户的登录信息,以方便站长实时了解自己网站的访问情况,选择了Apache 的log4j,但是在选择相对路径那块 卡主了,X度了好多方法(其实大多都是一样的内用,还一个字都不差的),都没有能解决问题,无奈搞了2天终于解决了,与大家分享一下
需求:
用户登录该网站时,把用户的登录名,ip,时间。统计到一个txt文档里,以方便其他系统调用此txt。项目名
- linux下mysql-5.6.23.tar.gz安装与配置
笑我痴狂
mysqllinuxunix
1.卸载系统默认的mysql
[root@localhost ~]# rpm -qa | grep mysql
mysql-libs-5.1.66-2.el6_3.x86_64
mysql-devel-5.1.66-2.el6_3.x86_64
mysql-5.1.66-2.el6_3.x86_64
[root@localhost ~]# rpm -e mysql-libs-5.1