背景
生物组织通常都是复杂的、由多种不同细胞类型组成。如血液包含淋巴系、髓系、粒系、红系等多种亚细胞类型;肿瘤组织也不仅仅是肿瘤细胞,也包含不同类型的非肿瘤细胞,如研究比较多的肿瘤浸润淋巴细胞(tumor-infiltrating lymphocytes )只是免疫细胞、基质细胞(stromal cells)的一部分,这种复杂的细胞组成也是通常所说的肿瘤微环境。因此,理解构成肿瘤微环境的细胞异质性是改善现有治疗的关键,也有利于发现新的预测性生物标志物,以及开发新的治疗策略。
单细胞技术虽然在解析组织细胞异质性中发挥着越来越重要的作用,但是对于临床大样本应用目前仍然面临挑战。通过计算的方法从表达矩阵分析混合物的细胞组成将会是有效的替代的方法。
常见的计算方法原理一种是基于去卷积(deconvolution)分解细胞组成比例,另一种是评估每种细胞的富集分值。
- 基于去卷积的方法有:
DeconRNASeq, CIBERSORT,UNDO等; - 基于富集分析的有:
xCell等;
1. DeconRNASeq: 基于RNA-Seq数据对组织样本去卷积
原文:“DeconRNASeq:A Statistical Framework for Deconvolution of Heterogeneous Tissue Samples Based on mRNA-Seq dat”
Bioinformatics 2013 Apr 15;29(8):1083-5. doi: 10.1093/bioinformatics/btt090.
DeconRNASeq 概述
DeconRNASeq 是一个R包,通过quadratic programming使用nonnegative decomposition algorithm对RNA-Seq数据分析,评估不同组织、细胞的混合比例。
需要两个个输入数据集:
- datasets : 需要分析的样本的表达矩阵(genes by samples)
datasets = signature *A - signatures : 参考数据集,即已知的特定细胞或组织的表达特征集合(genes by cell types)
- A : 参考数据集中每种细胞类型比例的矩阵(Cell type by samples)
DeconRNASeq 的使用方法
只要准备好了参考数据集和已知的细胞比例,DeconRNASeq的使用方法就很简单,但该方法的局限性也在于此,必需有合适的已知的参考数据集。
-
install deconRNASeq package
source("https://bioconductor.org/biocLite.R") biocLite("DeconRNASeq") library(DeconRNASeq) ##view documentation browseVignettes("DeconRNASeq") -
run the example
## multi_tissue: expression profiles for 10 mixing samples from multiple tissues data(multi_tissue) datasets <- x.data[,2:11] ## tissue-specific signatures for different human tissues signatures <- x.signature.filtered.optimal[,2:6] proportions <- fraction ## deconvolution DeconRNASeq(datasets, signatures, proportions, checksig=FALSE, known.prop = TRUE, use.scale = TRUE, fig = TRUE)-
datasets:
datasets matrix contains 28745 genes and 10 samples, column name are the sample names, row names are the gene names.
> head(datasets,3) reads.1.RPKM reads.2.RPKM reads.3.RPKM reads.4.RPKM reads.5.RPKM NR_024540 3.6682100 3.78953 8.254980 7.693440 5.637220 NR_028325.1 0.0796274 0.14644 0.104652 0.376109 0.104008 NR_028322.1 0.0796274 0.14644 0.104652 0.376109 0.104008 reads.6.RPKM reads.7.RPKM reads.8.RPKM reads.9.RPKM reads.10.RPKM NR_024540 6.358460 5.941820 6.555140 7.784240 5.9895300 NR_028325.1 0.160564 0.188188 0.133709 0.244789 0.0885794 NR_028322.1 0.160564 0.188188 0.133709 0.244789 0.0885794 > dim(datasets) [1] 28745 10 -
signatures:
The filter signature data matrix contains 1570 genes for the five tissues. Row names are the gene name, column names are the different tissue (or the cell type) in the mixture.
> head(signatures,3) brain muscle lung liver heart NR_024540 2.4742600 3.3782600 3.093570 1.279540 0.8652710 NR_028325.1 0.0675838 0.0556031 0.515925 0.085452 0.0830035 NR_028322.1 0.0675838 0.0556031 0.515925 0.085452 0.0830035 > dim(signatures) [1] 1570 5 -
proportions:
This data matrix means the proportions of different tissues(different cell types) in samples. Here is the prportions of 5 tissues in 10 sampes.
> head(proportions,3) brain muscle lung liver heart reads.1.RPKM 0.0463 0.0323 0.0805 0.0747 0.7662 reads.2.RPKM 0.0606 0.1156 0.0278 0.6960 0.1000 reads.3.RPKM 0.0728 0.6058 0.1051 0.1262 0.0900 > dim(proportions) [1] 10 5
-
2. CIBERSORT:基于SVR进行去卷积的网页版工具
原文:“Robust enumeration of cell subsets from tissue expression profile”
Nat Methods. 2015 May;12(5):453-7. doi: 10.1038/nmeth.3337.
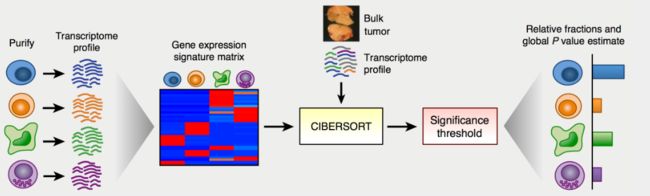
CIBERSORT 概述
CIBERSORT 是基于线性支持向量回归(linear support vector regression)的原理对人类白细胞亚型的表达矩阵进行去卷积的一个网页版工具。多用于芯片表达矩阵,对未知混合物和含有相近的细胞类型的表达矩阵的去卷积分析优于其他方法 (LLSR,LLSR,PERT,RLR,MMAD,DSA) 。该方法仍然是基于已知参考集,提供了22种白细胞亚型的基因表达特征集—LM22.
网址链接:http://cibersort.stanford.edu/
other six gene expression profiles deconvolution methods
- linear least-squares regression (LLSR),
- quadratic programming (QP),
- perturba- tion model for gene expression deconvolution (PERT),
- robust linear regression (RLR),
- microarray microdissection with analysis of differences (MMAD) ,
-
digital sorting algorithm (DSA)
 CIBERSORT
CIBERSORT
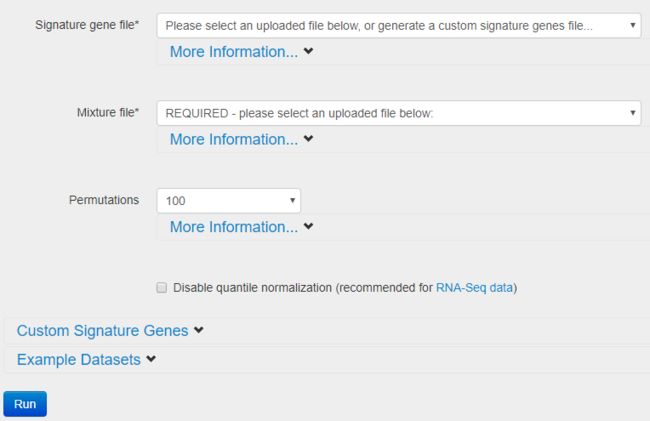
使用方法
- 首先在网页端注册账号
- 准备数据
- 需要分析的混合物样本
混合细胞表达矩阵,列是样本,行是基因; - 参考集
如果分析的样本适合用白细胞亚型的数据集,就不用自己准备,否则仍然需要准备参考数据集,即已知细胞类型的表达矩阵,行是基因,列是样本。
- 需要分析的混合物样本
- 上传数据,运行CIBERSORT
局限性
- 基于已知基因表达集,参考集仅包含白细胞亚型的特征基因表达集合。对于不适合该参考特征集的样本,需要准备参考集,但是不一定能找到合适的单细胞基因集合;
- 测试集是根据芯片数据的,并没有对测序数据测试验证
3. UNDO: 对肿瘤样本表达矩阵进行非监督去卷积
原文:“UNDO: a Bioconductor R package for unsupervised deconvolution of mixed gene expressions in tumor samples”
Bioinformatics. 2015 Jan 1;31(1):137-9. doi:10.1093/bioinformatics/btu607.
UNDO概述
UNDO(Unsupervised Deconvolution of Tumor-stromal Mixed Expressions)是针对肿瘤基质细胞混合表达矩阵非监督去卷积的一个R包,不需要已知参考集即可探索标记基因。该方法是根据表达非负性保证了标记基因的几何可识别性。首先检测位于混合表达的散射半径上的标记基因(只在肿瘤/基质中富集表达的基因),然后根据检测到的标记基因的表达值,通过标准化平均值估计细胞的比例,并对混合表达矩阵去卷积成肿瘤和基质的单个细胞群体的表达。
使用方法
输入文件只需要一个,即需要分析的混合表达矩阵,表达值是经过标准化后非对数转换的表达值。也可以输入已知比例的混合物矩阵,与最后计算的结果对比。
该R包包含5个函数:
- two_source_deconv:这个是主要的函数,可以调用出去卷积的其他亚函数。需要的输入数据是基因表达矩阵。
- gene_expression_input:这个函数是由two_source_deconv调用出的,用于检测输入的基因表达矩阵是否有效。
- dimension_reduction:
当样本超过2时,会进行主成分分析降维。 - marker_gene_selection:用于选择标记基因。
- mixing_matrix_computation: 基于marker_gene_selection的结果计算混合矩阵和纯化的矩阵水平。
- calc_E1: 当提供已知的混合矩阵比例时,用来计算E1值。如果E1值接近0时表明计算的结果与真实结果接近。
# install package
source("https://bioconductor.org/biocLite.R")
biocLite("UNDO")
library(UNDO)
browseVignettes("UNDO")
#load tumor stroma mixing tissue samples
data(NumericalMixMCF7HS27)
X <- NumericalMixMCF7HS27
# load mixing matrix for comparison
data(NumericalMixingMatrix)
A <- NumericalMixingMatrix
#load pure tumor stroma expressions
data(PureMCF7HS27)
S <- exprs(PureMCF7HS27)
two_source_deconv(X,lowper=0.4,highper=0.1,epsilon1=0.01, +epsilon2=0.01,A,S[,1],S[,2],return=0)
# compute the estimated pure source expressions
result <- two_source_deconv(X,lowper=0.4,highper=0.1,epsilon1=0.01, +epsilon2=0.01,A,S[,1],S[,2],return=1)
Sest <- result[[5]]
#draw the scatter plots between pure and estimated expressions of MCF7 and HS27
plot(S[,1],Sest[,1],main="MCF7" ,xlab="Estimated expression", ylab="Measured expression", xlim=c(0,15000), ylim=c(0,15000), pch=1, col="turquoise", cex=0.5)
plot(S[,2],Sest[,2],main="HS27" ,xlab="Estimated expression", ylab="Measured expression", xlim=c(0,15000), ylim=c(0,15000), pch=1, col="turquoise", cex=0.5)
4. xCell:富集分析混合表达矩阵细胞组成
目前对肿瘤微环境异质性去卷积的方法局限性有:
1)目前的方法依赖于纯化细胞类型的表达谱鉴定参考基因,因此数据来源对结果的可靠性影响非常大;
2)只关注肿瘤微环境很少一部分细胞类型,如多是免疫细胞类型,但是微环境中还包括血管细胞等多种细胞类型;
3)癌症细胞可以模仿表达免疫细胞特有的基因,但是只有很少的方法考虑到了这个因素;
4)大多方法没有得到全面证实,如通过细胞分选;
5)容易产生不同误差,因为严重依赖于预测的细胞类型;
6)预测相近细胞类型容易产生误差;
7)依赖参考文件矩阵的结构
xCell概述
- xCell是一种webtool和R软件包,可以从64种免疫和基质细胞类型的基因表达数据中进行细胞类型富集分析;
- xCell也是一种基于已知基因特征的方法,含有来自6种不同来源的数千种纯细胞类型;
- xCell应用一种新技术来减少紧密相关的细胞类型之间的关联。
- xCell的参考特征集使用广泛的计算机模拟和细胞计量学免疫分型来验证,并且显示出优于先前的方法。
- xCell允许研究人员可靠地描绘组织表达谱的细胞异质性景观
参考集数据来源:
参考基因表达集包括六个数据来源的RNA-seq和芯片表达数据
- the FANTOM5 project;
- the ENCODE project;
- the Blueprint project;
- the IRIS project;
- the Novershtern et al. study;
-
the Human Primary Cells Atlas (HPCA)
 gene signatue data sources
gene signatue data sources
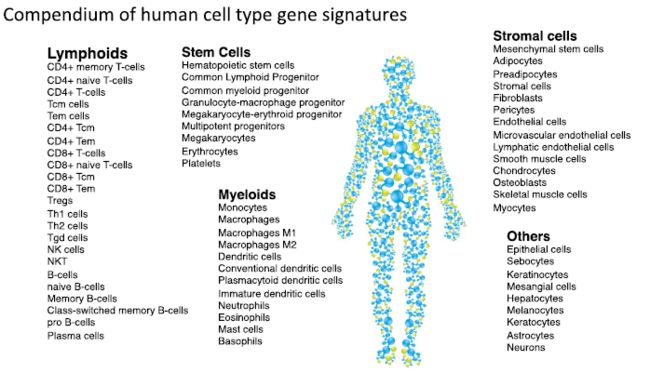
参考集包含的细胞类型:
包含64 种细胞类型, 有淋巴系亚型细胞, 干细胞亚型, 髓系细胞亚型, 基质细胞亚型 和其他细胞.
使用方法
xCell即提供了网页工具也提供了R包,需要的输入文件为混合基因表达矩阵。
- 准备混合表达矩阵
文件格式是tab分割的txt或csv,少于1gb,基因名为行名,样本名为列名,并且基因名需要时gene symbol,表达值是经过标准化的RPKM、FPKM或TPM。
GSM565269 GSM565270 GSM565271 ...
A1CF 5.9528 6.2118 6.0946
A2M 5.4145 5.4929 5.296
A4GALT 6.0914 5.7378 6.051
A4GNT 6.1141 6.0271 6.0217
AAAS 8.1466 7.8885 8.0305
AACS 6.703 6.4519 6.8497
...
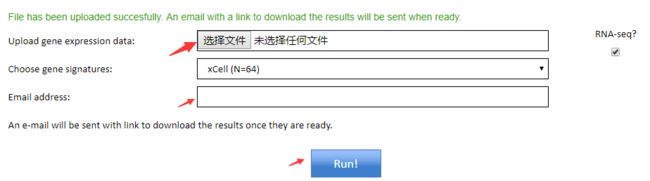
网页版分析
打开xCell网址 http://xcell.ucsf.edu/ ; 上传混合表达矩阵,写下邮箱,任务完成时可以从邮件下载结果。
基于 R 包
- Install the package
github: https://github.com/dviraran/xCell
source("https://bioconductor.org/biocLite.R")
biocLite("GSVA")
biocLite("GSEABase")
devtools::install_github('dviraran/xCell')
library(xCell)
- Run xCell
exprMatrix = read.table(expr_file,header=TRUE,row.names=1, as.is=TRUE)
xCellAnalysis(exprMatrix)
公布于2018—05.28
第 2 周 2018—05.28-06.03