欢迎关注”生信修炼手册”!
Seurat是一个分析单细胞转录组数据的R包,提供了t-SNE降维分析,聚类分析,mark基因识别等多种功能,网址如下
https://satijalab.org/seurat/
基本用法如下
1. 导入10X 单细胞数据
library(Seurat)
input_dir <- "/scRNA/outs/filtered_gene_bc_matrices/GRCh38/"
pbmc.data <- Read10X(data.dir = input_dir)
pbmc <- CreateSeuratObject(raw.data = pbmc.data, project = "10X")2. 预处理
预处理就是根据基因的表达量等特征,对细胞进行过滤,通常的做法就是指定一个阈值,比如要求一个细胞中检测到的基因数必须大于100,才可以进入到下游分析,如果小于这个数字,就过滤掉该细胞。
需要强调的是,预处理这一步是可选的,在设定过滤的阈值时,需要人为判断,这样的设定方式会受到主观因素的干扰,所以往往都会指定一个非常小的过滤范围,保证只过滤掉极少数的离群值点。
为了指定一个合适的阈值,我们首先需要查看细胞中不同特征的分布,常见的有以下几个指标
1.nGene
2.nUMI
3.mito.percentnGene代表的是在该细胞中共检测到的表达量大于0的基因个数,nUMI代表的是该细胞中所有基因的表达量之和,mito.percent代表的是线粒体基因表达量的百分比,通过小提琴图来展示对应的分布,代码如下
# 提取线粒体基因列表
mito.genes <- grep(
pattern = "^MT-",
x = rownames(x = pbmc@data),
value = TRUE)
# 统计线粒体基因丰度的百分比
percent.mito <- Matrix::colSums([email protected][mito.genes, ]) / Matrix::colSums([email protected])
# 将统计的百分比数据添加对象中
pbmc <- AddMetaData(
object = pbmc,
metadata = percent.mito,
col.name = "percent.mito")
# 小提琴图
VlnPlot(
object = pbmc,
features.plot = c("nGene", "nUMI", "percent.mito"),
nCol = 3)生成的图片示意如下
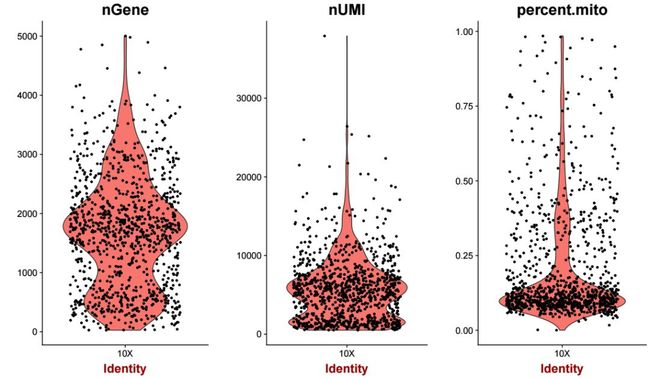
图中每个点代表的是一个细胞,反应的是对应特征在所有细胞的一个分布情况。通过观察上图,我们可以确定一个大概的筛选范围。以nGene为例,可以看到数值在4000以上的细胞是非常少的,可以看做是离群值,所以在筛选时,如果一个细胞中检测到的基因个数大于4000,就可以进行过滤。
在过滤阈值时,我们还需要考虑一个因素,就是这3个指标之间的相互关系,可以通过以下方式得到
par(mfrow = c(1, 2))
GenePlot(object = pbmc, gene1 = "nUMI", gene2 = "percent.mito")
GenePlot(object = pbmc, gene1 = "nUMI", gene2 = "nGene")结果示意如下
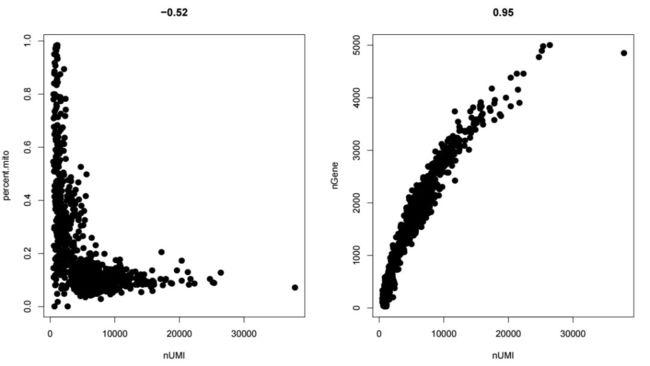
以UMI和gene之间的关系图为例,可以看到非常明显的一个相关性,当gene个数为4000时对应的umi在20000左右,所以在设定阈值,我们想过滤掉gene大于4000的细胞,此时umi的阈值就应该设置在20000左右。
结合以上两种图表,判断出一个合适的阈值之后,就可以进行过滤了,代码如下
pbmc <- FilterCells(
object = pbmc,
subset.names = c("nGene", "percent.mito"),
low.thresholds = c(200, -Inf),
high.thresholds = c(2500, 0.05))subset指定对应的特征,low和high分别指定合理取值的两个范围。
3. 归一化
预处理之后,首先进行归一化,代码如下
pbmc <- NormalizeData(
object = pbmc,
normalization.method = "LogNormalize",
scale.factor = 10000)默认采用LogNormalize归一化算法,首先将原始的表达量转换成相对丰度,然后乘以scale.factor的值,在取对数。
归一化之后,Seurat提取那些在细胞间变异系数较大的基因用于下游分析,代码如下
pbmc <- FindVariableGenes(
object = pbmc,
mean.function = ExpMean,
dispersion.function = LogVMR,
x.low.cutoff = 0.0125,
x.high.cutoff = 3,
y.cutoff = 0.5)在进行下游的降维和聚类分析之前,有必要进一步降低系统误差,代码如下
pbmc <- ScaleData(
object = pbmc,
vars.to.regress = c("nUMI", "percent.mito")
)3. 聚类分析
聚类分析用于识别细胞亚型,在Seurat中,不是直接对所有细胞进行聚类分析,而是首先进行PCA主成分分析,然后挑选贡献量最大的几个主成分,用挑选出的主成分的值来进行聚类分析。
第一步,PCA分析,代码如下
pbmc <- RunPCA(
object = pbmc,
pc.genes = [email protected])第二步,评估最显著的主成分,代码如下
pbmc <- JackStraw(
object = pbmc,
num.replicate = 100,
display.progress = FALSE)这一步特别费时,运行完之后,可以进行如下可视化
JackStrawPlot(object = pbmc, PCs = 1:12)
数值小于0.05的,认为是显著的,从上图可以看出,PC1-9是最显著的主成分。
第三步,聚类分析,代码如下
pbmc <- FindClusters(
object = pbmc,
reduction.type = "pca",
dims.use = 1:9,
resolution = 0.6,
print.output = 0,
save.SNN = TRUE)聚类的结果通过以下代码可以查看
pbmc@ident4. t-SNE降维分析
采用t-SNE降维算法,用2d-plot的方式展示细胞的表达量分布,同时根据聚类结果,将同一cluster的细胞用相同颜色表示,代码如下
pbmc <- RunTSNE(
object = pbmc,
dims.use = 1:10,
do.fast = TRUE)
TSNEPlot(object = pbmc)生成的图片如下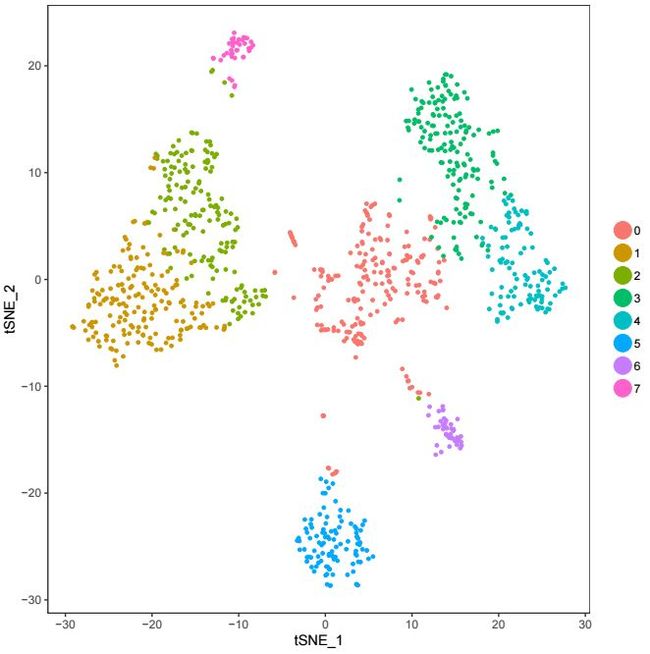
还支持在图上标注每个cluster的名字,代码如下
current.cluster.ids <- c(0, 1, 2, 3, 4, 5, 6, 7)
new.cluster.ids <- c(
"CD4 T cells",
"CD14+ Monocytes",
"B cells",
"CD8 T cells",
"FCGR3A+ Monocytes",
"NK cells",
"Dendritic cells",
"Megakaryocytes")
pbmc@ident <- plyr::mapvalues(
x = pbmc@ident,
from = current.cluster.ids,
to = new.cluster.ids)
TSNEPlot(
object = pbmc,
do.label = TRUE,
pt.size = 0.5)
5. mark基因识别
通过差异分析来识别每个cluster下的标记基因,将该cluster下的细胞作为一组,其他cluster下的细胞作为另一组,然后进行差异分析,代码如下
> all_markers <- FindAllMarkers(object = pbmc)
> head(all_markers)
p_val avg_logFC pct.1 pct.2 p_val_adj cluster gene
UBA52 1.344025e-52 -1.133346 0.327 0.919 4.507589e-48 0 UBA52
RPL39 1.708604e-52 -1.241172 0.398 0.924 5.730315e-48 0 RPL39
MT-ATP6 7.859458e-51 1.092949 0.995 1.000 2.635905e-46 0 MT-ATP6
RPS24 4.304581e-50 -1.116990 0.445 0.934 1.443670e-45 0 RPS24
TPT1 1.552635e-49 -1.106253 0.611 0.965 5.207229e-45 0 TPT1
MT-CYB 2.423599e-49 1.125433 0.991 0.999 8.128265e-45 0 MT-CYB同时提供了基因在不同cluster下的小提琴图,代码如下
VlnPlot(
object = pbmc,
features.plot = "UBA52")生成的图片如下
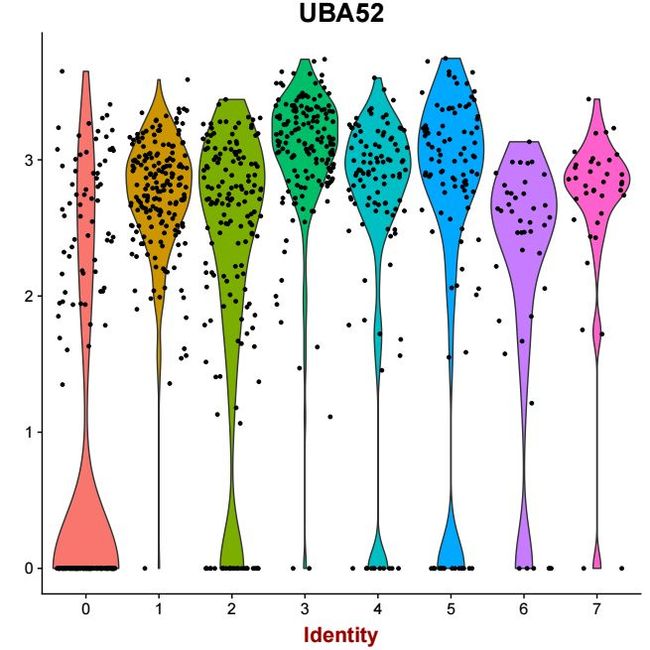
更多的用法请参考官方文档。
·end·
—如果喜欢,快分享给你的朋友们吧—
扫描关注微信号,更多精彩内容等着你!
