今天将会用到第二天建立的rnaseq环境及对应文件路径和Day3下载好的.sra数据
今天主要学习内容是配置相关软件环境,开始数据转换,质量控制,了解参考基因组及注释文件
数据转换:其实这一步就是将下载好的.sra文件转换成fq文件,因为研究者需要将下机的fq文件压缩成sra上传到ncbi,我们这一步的工作就是再将其还原。主要用到的工具是fastq-dump,它和Day3天的perfetch同样数据sratools工具包,所以直接调用就可以。
数据质控:这一步的目的就是通过查看有无接头和低质量碱基等,得知原始数据质量咋样,不好就要进行处理,处理之后再不好那可能就是要重新检测,主要用到的软件是fastqc和multiqc,其中当我们分析数据存在多个样本时,为了方便同时查看质量信息我们将使用multiqc进行多样本合并呈现结果。
数据过滤:通过数据质控知道了数据所存在的问题,那么接下来我们要开始对原始数据进行过滤,因为接头盒低质量的碱基都会影响后续的比对、定量准确性,及下游分析。主要用到的工具为trim_galore或者trimmomatic,SOAPfilter(这三个区别)
比对:比对的软件有很多,主要有三种类型:基于基因组比对:star,hisat2;基于转录组比对:bowtie,bwa;不基于比对:salmon
定量:推荐使用featureCounts,它是subread软件下的一个小软件
开始!
第一步-软件环境
依然是使用conda进行配置,主要软件包括:数据转换,质量控制,数据过滤,比对和定量
#激活之前设置好的rnaseq环境
conda activate rnaseq 或 source activate rnaseq
#安装所需软件; -y:默认安装过程全部yes,当conda把所有软件都安装好之后才会给反馈
conda install fastqc multiqc trim-galore trimmomatic hisat2 bowtie2 subread salmon -y
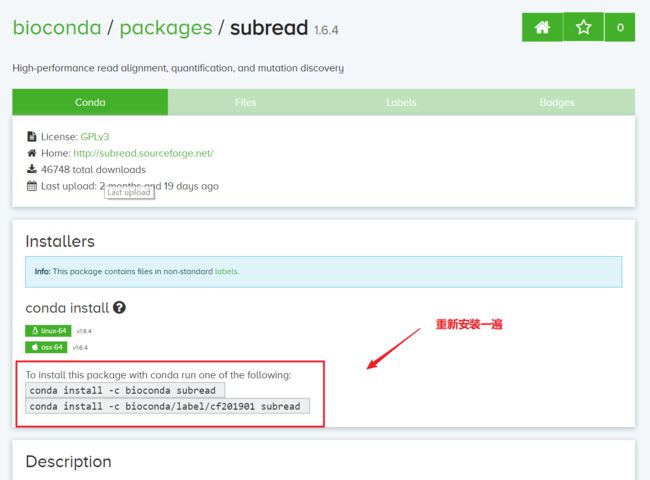
请注意软件名称大小写,很重要,错了容易报错;同时还有可能存在软件依赖性,比如这里的featurecounts找不到并报错,此处的意思是找遍了所有conda下载channel也没找到它,这是我们就网页搜索一下,根据正确答案将代码替换就ok啦
当我将fastqc multiqc trim-galore trimmomatic hisat2 bowtie2 subread salmon这些软件都安装好后,每一个软件我都检测一下到底安装好没,通过:输入软件名直接回车、软件名 --help、软件名 -h、软件名 -help,以上方法均为查看该软件帮助文件,当帮助文件显示后则表示为安装成功。
但!请看(学习不认真就要入深坑)
这两个软件没有显示帮助文件,可能是安装出了问题,那么我通过网页搜索其安装方法,进行重新安装,如下,但还是出现问题:
问题一:
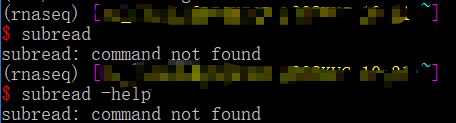
我以为的真的不是我以为的,我以为所有的软件的帮助文件都是这样看的,结果我错了,首先subread不是单独的软件,它还包括subread-align,subjunc,其官网:( http://subread.sourceforge.net/),下这个软件的目的是为了使用其下面的featureCounts,所以我们直接输入 featureCounts回车,也可以证明我们这个软件安装成功
问题二:
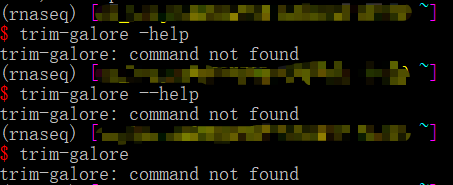
trim-galore也不显示帮助文件,也重新安装了,没报错,去安装的路径(home/miniconda3/envs/rnaseq/bin)下看它也在里面呀,那这又咋了啊!真让人头大!
我的天!这是我们可以vi ~/.bashrc,将其添加到变量中export PATH=~/miniconda3/envs/rnaseq/bin:$PATH,source ~/.bashrc激活一下,只是再trim_galore回车就会显示帮助文件信息了
所以基于以上,当我们遇到安装软件之后不显示帮助文件信息时处理方式有三种(目前我遇到的,谁知道以后还会遇见啥呢)
1.搜索conda 软件名:根据网站提示重新安装
2.通过1没有报错,安装成功,但还是不显示帮助文件,那我们就要了解这个软件是不是唯一的,其内部会不会还有其他软件(类似问题一)
3.查看软件安装路径内是否有该软件,有的话直接添加变量就OK了(类似问题二)
Ok!配置相关软件环境完成
第二步-数据转换
由于目前测序都是双末端测序,因此将sra转换成fq后会出现两个fq文件,read1和read2,将下列脚本写入名字为sra2fq.sh中(vi sra2fq.sh)
cat srr.ids | while read I ;do
echo $i
#time fastq-dump –gzip –split-3 -A $i 路径/${i}.sra -O 输出路径 &;
done
首先在做循环脚本时,真正要运行的命令先用#注释掉(小泽提的好建议),用echo $i可以先看看这个循环是否是可行的,运行一下,看结果:
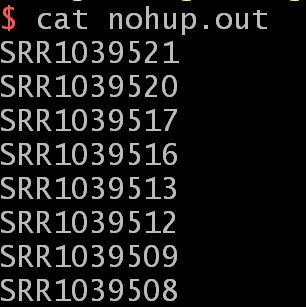
确认是我们要分析的数据列表,好了,这时回到脚本将注释打开
当我们在使用某一个软件时,帮助软件时非常有用的,因为很多参数我们没有必要都要记住,可能也记不住,所以我们要充分利用好帮助信息,查看某个软件的帮助信息的命令前面已经说了太多了(-help)
如何可以让一个安装好的软件被我们所用,也就是将它运行起来,这里用featureCounts举例(因为我终于弄明白了subread的帮助文件怎么显示了);主要三因素,主程序+选项+参数:featureCounts [option] -a
,[]里内容表示可选,<>里内容为必选选项,不可忽略,在写命令时是不需要写上[]和<>。
此外,在帮助文件中你会看到有一个连字符+大/小写字母,和两个连字符+单词,两个连字符+单词要比一个连字符+大/小写字母说的更详细。这两种方式主要是对选项的调用,后面才是选项的详细描述内容。
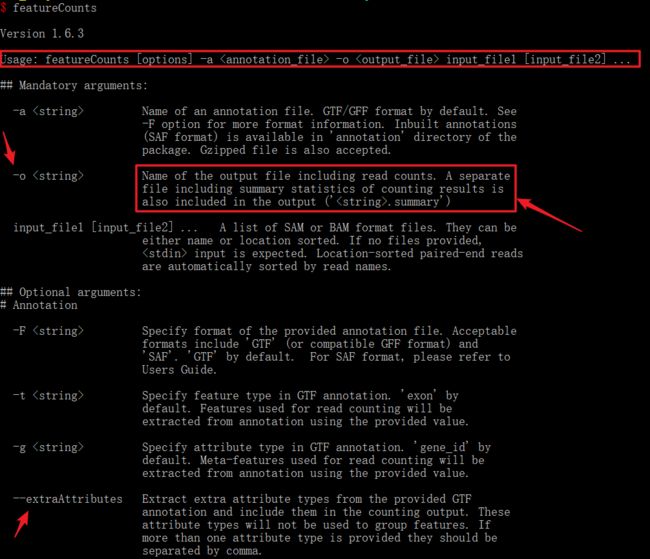
数据转换命令详述:
time fastq-dump –gzip –split-3 -A $i 路径/${i}.sra -O 输出路径 1>sra2fq.log 2>&1 &
#time:表示计时,就是转换这个过程用了多久,可有可无
#fastq-dump:主程序
#--gzip:表示将解压出来的fq还是压缩成gzip格式,目的是节省空间内存
#--split-3:https://www.biostars.org/p/156909/,虽然其中有个3,但其实结果一般也就出2个文件
#-A指定输出结果名
#i:SRRxxxx
#-O:指出输出文件路径
#1>sra2fq.log:将结果的正确日志文件输出到sra2fq.log
#2>&1:2表示将错误信息,并将其追加到1的正确日志中。也可以将2错误信息丢弃到linux黑洞中去:2>/dev/null
#&:实现并行运算的意思,说白了就是同时跑这个循环,对i进行同时数据转换
--split-3说白了就是输出3个结果文件如下,但是我们往下分析的时候只用read1和read2,所以如果只想要2个结果文件,可以用--split-files;

好了,现在可以投任务了 nohup sh sra2fq.sh &,投任务这个时间可以准备下载参考基因组和注释文件了
第三步-参考文件
为什么需要参考文件?都有哪些参考文件?
第一个参考文件:参考基因组
转录组最终的结果就是要比较表达量的差异,那么一堆随机排列的碱基怎么看表达量差异呢?就需要一个“标准”,我们测到的不同数据都和这个标准去做比较,比上的多,我们就认为表达量高,相反表达量少,对于比对不上的,我们认为不表达。其实这里说的“标准”也就是第一个参考文件指的就是参考基因组,人类基因组经历了从hg16、hg17、hg18、hg19、hg38版本,最新版本即为hg38。
人类参考基因组:gz压缩文件800多M,解压后3G。
有3种主流的数据库可供使用下载不同物种基因组数据:NCBI、UCSC、Ensembl;
-NCBI:ftp://ftp.ncbi.nlm.nih.gov/genomes/
-UCSC:http://hgdownload.cse.ucsc.edu/downloads.html
-Ensembl:ftp://ftp.ensembl.org/pub/
第二个参考文件:基因组注释文件
有了基因组,可以让我们去探索得到的数据reads中哪些序列匹配到基因组上的对应位置,但是我们还是不知道对应的是什么,如果我们把基因组比作“寻路”的过程,那么注释文件就是给找好的路指定“路牌”,主要使用两个数据库:Gencode:Ensembl,下载得到的文件主要是GTF和GFF:
Gencode:https://www.gencodegenes.org/,包括人和小鼠的
Ensembl:ftp://ftp.ensembl.org/pub/current_gtf
关于GTF和GFF文件的信息请学习:小泽分享的GTF与GFF的小趣事
那么现在开始下载两个参考文件吧!
请将两个参考文件下载到之前创建的ref文件目录下(cd ref)
#genome(从ensembl下载)
wget -c ftp://ftp.ensembl.org/pub/current_fasta/homo_sapiens/dna/Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
#annotation(从ensembl下载)
wget -c ftp://ftp.ensembl.org/pub/current_gtf/homo_sapiens/Homo_sapiens.GRCh38.96.gtf.gz
# -c的含义是断点续传
# 如果下载速度慢,可以放后台
#解压
gzip -d Homo_sapiens.GRCh38.96.gtf.gz
gzip -d Homo_sapiens.GRCh38.dna.primary_assembly.fa.gz
第四步-利用fastqc进行质控
通过fastq-dump将.sra文件转换成fq后,共生成8对SRRxxxxxx_1.fastq.gz和SRRxxxxxx_2.fastq.gz,接下来对其进行fastqc质控,在qc文件目录下进行(cd qc)
fastqc 路径/raw/*.gz -o ./ -t 10
# -t --threads 选择程序运行的线程数,越多越快哦;-o输出路径;*.gz表示该目录下所有的.gz文件;
因为我的fastq-dump命令还没有跑完,所以我单独跑已完成的.fastq.gz(此处我跑两个SRRxxxxx数据文件,方便后面multiqc使用及结果演示),如下:
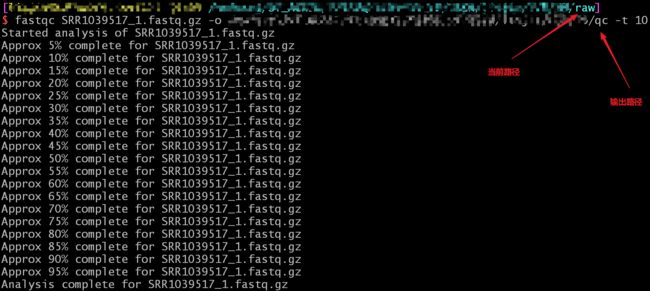

运行结果会按照每个fq文件以html格式输出,然后利用filezilla导出结果(我用的导出结果是WinSCP)如下:
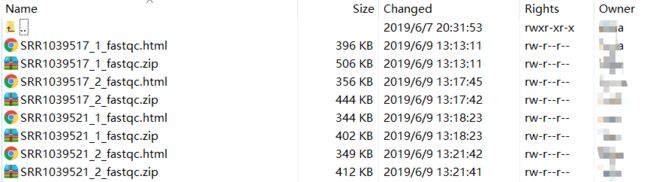
右键.html即可查看结果,如下:
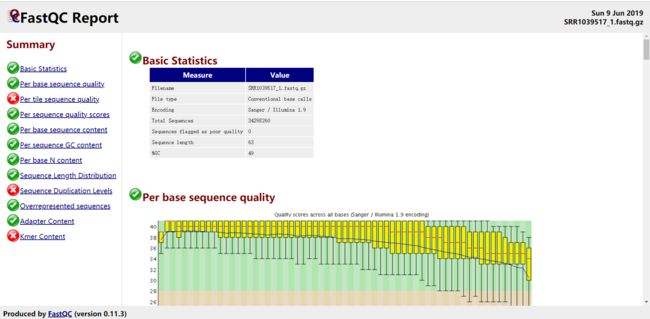
至于这个Report怎么解读,请学习 继续小泽的优秀文章====不要"太重视“fastqc的结果
FastQC Report详解: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/Help/
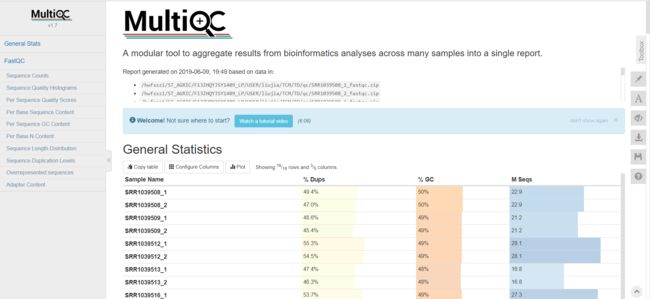
因为我们最终会输出16个.html,一个一个看实在是太麻烦,所以我们利用multiqc可以将结果合并呈现
额外说明一下multiqc安装有两种方法:官网:https://multiqc.info/docs/
# pip安装
pip install git+https://github.com/ewels/MultiQC.git #Installation with pip
# conda安装
conda install -c bioconda multiqc # Installing with conda
插播:在等待安装multiqc时,所有数据fastqc结束,如下;
运行MultiQC
直接指定MultiQC要分析的文件路径即可,若数据在当前目录下输入multiqc .即可。
multiqc .
#使用--ignore忽略掉某些文件
#multiqc . --ignore *_R2*
multiqc结果解析,请学习:https://www.jianshu.com/p/f83626fd1fa1
坑!坑!关于multiqc
我以为的又不是我以为的,2个小时出坑
一定要清楚multiqc是干啥的,我一直以为是和fastqc一样的进行质控,错!我以为错了,其实它是将fastqc质检后生成的.zip文件进行质量信息结果合并呈现,所以我们应该在qc这个路径下对.zip文件进行multiqc .
顿悟这个问题的心里路程:
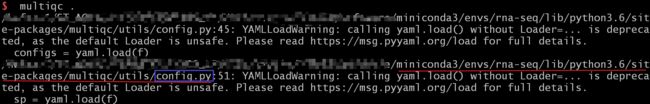
当我第一次运行multiqc .最先出现这个报错,下面继续出错显示WARNING
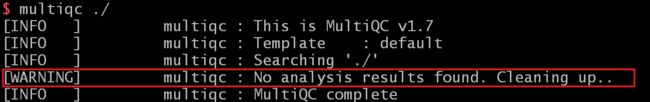
顿时蒙圈了,别人也没这个情况呀,我就开始搜索,搜呀搜呀搜呀搜,第一个警告解决,帮助来自 https://github.com/yaml/pyyaml/wiki/PyYAML-yaml.load(input)-Deprecation#footnotes
找到config.py文件,对其进行编辑,vi 路径/config.py
在vi编辑器中查找yaml.load,一般都是yaml.load(f),这是我们要在其后面加上Loader=yaml.FullLoader,把所有的yaml.load中都加上,之后保存wq,退出vi
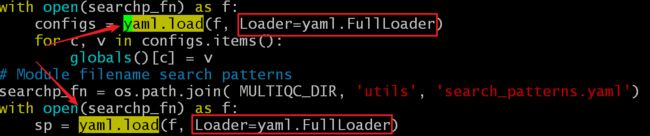 yaml.load (f, Loader=yaml.FullLoader)
yaml.load (f, Loader=yaml.FullLoader)
接下来我们开始解决WARNING,用了1个小时解决的,我都想爆粗口,现在想想就一句话,multiqc处理的是fastqc质控后生成的.zip,找对路径找对文件执行就完事了,要被自己蠢哭了,最后生成multiqc_data和.html,使用WinSCP打开.html