在找到很多变异以后(本文具体指的是snv以及indel),我们需要做的就是进行注释了,这是因为无论WGS还是WES,都会call出大量的变异。即使是一个正常人,也会有百万数量级的变异(详见上一章),而我们需要做的是把其中可能有害的变异找出来。因此对于这么多的变异,就需要通过注释信息来筛选了(不同于call snv/indel以后的筛选,那是依据变异本身的可信度进行筛选的)。
对于不同的研究目的,目前用的注释软件还是不少的,有专门针对肿瘤数据的注释软件(如oncotator等),也有孟德尔遗传疾病测序数据常用的annovar、vep等。
本文主要讲的是通过annovar,对vcf文件进行注释,个人觉得annovar特别好用简单,说明书也写得很好,即使是我这种看了英语头晕的人,也能看的比较明白。
比较一下annovar和vep,annovar软件是需要先申请再下载的,对于研究机构是免费的,对商业机构是收费的,只要你拿学邮去申请,应该就没有问题。而vep是免费的,但是呢,安装起来超级麻烦,本文虽然不用vep,但会在后面附上我当初安装vep的笔记。
annovar很人性化的一点是,当你下载到软件以后,直接解压就能用了,不用安装。而你需要注释什么数据库,按照官网说明书的词条直接下载就行。
本文主要讲述我在做课题的时候是如何使用的,具体的细节可以参考官网说明书或者别的大神们的使用笔记。
首先是下载数据,annovar提供了大量的数据库,可以单独下载也可以批量下载,主要是来源于annovar自己网站以及ucsc的,你要是批量下载是完全没有必要的,因为一个数据库可能有很多的版本,也有很多数据库是你不需要的,还有一些数据库所占空间那是相当的大。举几个我常用的数据库为例(我只是举例一些,建议是看看说明书根据需要下载)。
#注释基因
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar refGene humandb/
#注释人群频率
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar esp6500siv2_all humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar exac03 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar gnomad_genome humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar 1000g2015aug humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar popfreq_max_20150413 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar popfreq_all_20150413 humandb/
#预测打分
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar mcap13 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar revel humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar gerp++gt2 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar cadd13 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar dbnsfp35a humandb/
# dbsnp
$ perl annotate_variation.pl -downdb -buildver hg19 snp151 humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar avsnp150 humandb/
# 致病位点公共数据库
$ perl annotate_variation.pl -downdb -buildver hg19 gwasCatalog humandb/
$ perl annotate_variation.pl -downdb -buildver hg19 -webfrom annovar clinvar_20190305 humandb/
#区域注释
$ perl annotate_variation.pl -downdb -buildver hg19 cytoBand humandb/
# 稍微注意一下 humandb/ 是当前目录下有这么一个文件夹,用来放数据库的,这个路径关系到你用的时候的路径,你放在哪,用的时候把路径改成哪就行
然后是输入格式的转换,对于注释所需的输入文件,只需要前五列按照avinput的要求就行了,格式如下。
#分别以tab分隔
染色体位置 起始位点 终止位点 参考基因组碱基 突变碱基
在注释完以后是不会保留原有的五列以外的内容的,转换脚本如下:
$ perl convert2annovar.pl -format vcf4old in.vcf > out.avinput
上面这个脚本除了把vcf转换为avinput以外,还会把一些多等位位点转换为单等位位点来注释,而格式之所以选用vcf4old,是因为我的输入格式是家系merge的vcf,一个vcf文件里有一家人的信息,如果是单个样本的话,直接用vcf4就行了。
用这种方式得到的结果就像我说的,注释完以后就没有保留 genotype等信息,除了前五列就只剩注释信息了。当然,也可以不转换格式,直接用vcf作为输入格式了,那么注释结果以标签的方式插入vcf文件之中,但是这样的话,会导致输出结果异常的占空间,因为,每一个数据库的头文件会反复出现在每一行被注释的变异中。提供参考脚本如下:
# 输入是vcf文件,要加上参数-vcfinput
$ perl table_annovar.pl in.vcf /path/humandb/ -buildver hg19 -out myanno -remove -protocol refGene,cytoBand,exac03,avsnp147,dbnsfp30a -operation g,r,f,f,f -nastring . -vcfinput
虽然提供了这种方法,但我却是不建议的,尤其是我以WGS的文件作为输入,会使输出文件占很大的空间,但我又想保留 genotype信息,因为后面我需要这个信息判断变异的显隐性,这对于筛选是很关键的,那怎么办呢?
我想到的办法就是,在转换格式的时候,添加参数-includeinfo和-comment,前者的作用保留了genotype信息,后者保留了vcf的头文件。
$ perl convert2annovar.pl -format vcf4old in.vcf -outfile out.avinput -comment -includeinfo
转换完格式就是注释了,虽然annovar提供了多种注释方式,但是我们一般都是批量注释,即像上面那个注释的例子一样,下面来简单说一下怎么批量注释。
$ table_annovar.pl out.avinput /your/path/of/humandb/ \
--buildver hg19 -out /your/path/of/outfile -remove \
-protocol refGene,cytoBand,EAS.sites.2015_08,ALL.sites.2015_08,kaviar_20150923,hrcr1,cg69,gnomad_genome,exac03,exac03nonpsych,esp6500siv2_all,cg46 \
-operation g,r,f,f,f,f,f,f,f,f,f,f \
-nastring . --thread 12
来详细说一下我这个脚本有什么需要注意的地方,而我又是怎么应用于实际课题中的。
首先是out.avinput,就是上面转换之后的输出文件;之后跟着的就是我们下载数据库的目录;我用的是hg19的参考基因组;-out是输出文件的路径和输出文件的前缀;-remove表示删除注释过程中的临时文件;-protocol表示注释使用的数据库,用逗号隔开;-operation 表示对应顺序的数据库的类型(g代表gene-based、r代表region-based、f代表filter-based,可对照说明书),用逗号隔开,与-protocol顺序一一对应;-nastring 表示用点号替代缺省的值。
annovar其实提供了好多个gene-based数据库下载,refGene是NCBI提供的,还有ensembl、UCSC提供的ensGene、knowGene,这三个数据库是有所差别的,至于怎么选择,建议就和使用的参考基因组来源一致即可,比如我用的是NCBI上下载的参考基因组,那么就用refGene。
-csvout 表示最后输出.csv文件,使用这个参数得到的是csv格式的文件,可以直接用excel打开,但是我没有使用,输出是tab分隔的txt文件,方便后续我写脚本处理。为什么“,”分隔的csv文件不适合处理呢?那是因为refgene的注释结果也是逗号分隔的,在分析的时候就会出现问题。
--thread是线程数,当然线程数越高注释速度越快。
到了这一步以后,其实结果还是前五列avinput的内容,后面是注释的内容,genotype的信息还是没有掉了,这个时候就加一步,来解决一下这个问题。
$ grep -v "##" out.avinput | cut -f1-9 --complement > gt.txt
$ paste hg19_multianno.txt gt.txt > merge.anno.txt
很简单的两行命令,第一行提取了avinput文件里的genotype信息,然后第二行merge这个信息到注释完的文件上。这样得到的结果呢,又有注释信息,又有genotype信息,就很方便进行trio分析。
到这里其实就差不多了,可以进行下一步的筛选了,但是实际情况中,我们用到的数据库可能不止annovar提供的这些。毕竟annovar提供的数据库都是公开的数据库,而且对于日新月异的预测数据库等,annovar作者也不能做到每一种都去收录,如果你有一些自己下载的数据库,你该怎么利用annovar去注释呢?
这一类的数据库很多,比如omim需要申请才能下载,HGMD专业版需要付费订阅,这些annovar都是没有提供的。但是这些数据库在孟德尔遗传疾病中是很具有参考价值的,由于这两个都不能直接下载获得,这里也不详述,以两个近期发表在Nature Genetic上的公开的预测数据库为例,简述一下简单的方法构建自己的数据库。
annovar注释变异有两种注释方式,一个是基于区域,一个是基于位点,CCRS就是一个基于区域的有害性预测打分工具,其数据可在这里下载,分为常染色体和X染色体;S-CAP就是一个基于位点的splicing 区域有害性预测工具,其数据可以在这里下载。
CCRS
由于是基于区域的,那么在-operation参数里面呢是r,然后呢从网上下载下来的是一个bed文件,截取前十行,长这样。
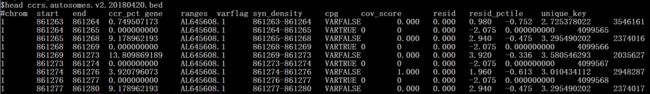
这些信息其实我们只需要ccr_pct这一列的打分,为了方便起见,完全可以只保留前四列的内容,不然注释完会把所有前三列以后的内容都加到注释结果文件里。
那么这么一个文件是不能直接拿来用的,第一步就是改名。下载过annovar提供的数据库后可以知道,annovar使用的数据库都有自己的命名方式,这里的话直接改成 hg19_ccrs.txt即可,hg19表示所使用的参考基因组版本,因为CCRS 的坐标是基于hg19的,所以这里是hg19,ccrs就是-protocol参数里的标识符了,然后以.txt作为后缀。
然后是修改格式,一开始我用的方法是参考annovar提供的region-based数据库的格式,然后修改CCRS的格式。后来我发现这是行不通的,因为现成的数据库格式差别有点大,那么就只能看源码了。但其实我是不会perl的,在师姐的帮助下,完美的解决了这个问题。
虽然我们是使用table_annovar.pl进行注释的,实际上对于region-based数据库的注释是调用annotate_variation.pl,所以需要修改annotate_variation.pl的源码。在2998行我们可以看到,其实对于不同region-based数据库,都是有预设的。
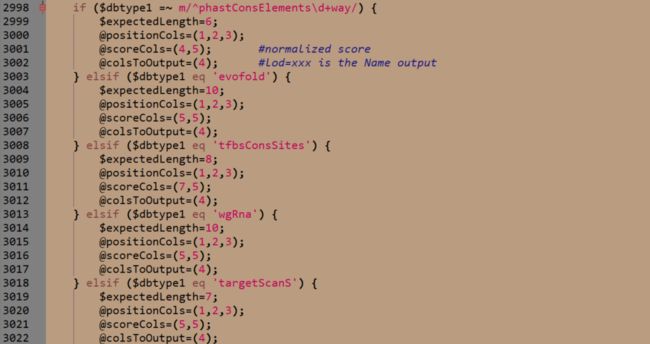
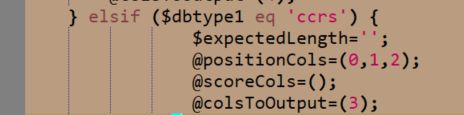
当然也可以根据最后一个else语句块修改CCRS的格式
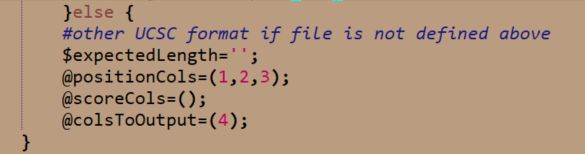
从这个语句块可以看出来,只需要在CCRS原始文件前面添加一列内容,以tab隔开,就行,添加啥也都无所谓,这里就不赘述了。
S-CAP
由于是基于位点的,那么在-operation参数里面呢是f,然后呢从网上下载下来的文件,一共有八个文件(不同区域不同标准,可以参考论文),任意选取一个截取前十行,长这样。
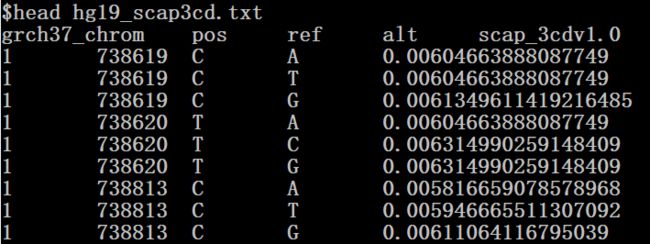
这里我已经把名字修改了,如果大家有查看过filter-based数据库,大抵都是这个格式,染色染位置、起始位置、结束位置、参考碱基、突变碱基、要注释的内容(可以是多列),以tab隔开。这里可以发现,少了一列结束位置,那是因为论文只针对snp,所以不用start、end表示也可以,这里用一个简单的语句就可以解决这一点。
awk -F "\t" '{print $1"\t"$2"\t"$2"\t"$3"\t"$4"\t"$5}' scap3cr.txt > hg19_scap3cr.txt
但是注意,这样是不够的,如果你看了这个论文,你会发现,这是预测splicing区域有害性的打分,更重要的是,论文中对splicing区域的划分和annovar默认的aplicing区域是不一样的!!!
在annovar里头,splicing的区域是2bp,也就是图中的1和4.
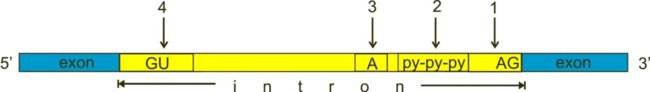
但是论文中作者为了更好的预测有害性,用了整整100bp的长度(内含子外显子各50bp),所以仅仅用annovar默认的splicing区域大小就太浪费这个打分了,那怎么扩大这个范围呢?
其实如果仔细看annovar说明书我们会发现,在
annotate_variation.pl的脚本中有一个参数
--splicing_threshold,可以控制splicing的区域大小,默认是2,但是我们用的是
table_annovar.pl的脚本进行注释,虽然是调用
annotate_variation.pl,但是却没有了
--splicing_threshold这个参数,看来又只能改源码了。
在
table_annovar.pl第380行(不同版本可能不太一样),有这么一句话。
里面在调用annotate_variation.pl的时候,直接给了--splicing_threshold这个参数,只要把后面的数字改成50就好了(原来是2)。至于怎么建立索引可以参考这里,是否建立索引会影响注释速度,但不会影响结果。
至此,这一部分内容就完了,我在实践中也就是这么使用的,具体的根据需要可以好好读一读annovar的官网说明。
水平有限,要是存在什么错误请评论指出!请大家多多批评指正,相互交流,共同成长,谢谢!!!
附(vep安装笔记)
#由于我用的是ubuntu server 18.04,不同linux发行版本之间的安装可能存在差异
$ git clone https://github.com/Ensembl/ensembl-vep.git
$ cd ensembl-vep
$ perl -MCPAN -e shell
$ cpan>install DBI
$ cpan>install Bio::DB::HTS
$ cpan>install LWP::Simple
$ cpan>install LWP::Protocol::https
$ cpan>install Archive::Extract
$ cpan>install Archive::Tar
$ cpan>install Archive::Zip
$ cpan>install CGI
$ cpan>install Time::HiRes
$ cpan>install DBD::mysql
$ cpan>q
$ sudo apt-get install libdbd-mysql-perl
$ perl INSTALL.pl
$ perl INSTALL.pl --AUTO af --SPECIES homo_sapiens --ASSEMBLY GRCh37 --DESTDIR /your/path/of/vepdatabase/ --CACHEDIR /your/path/of/vepdatabase/
$ perl convert_cache.pl --species homo_sapiens --version 94_GRCh37 --dir /your/path/of/vepdatabase/