什么是illumina bcl 格式和fastq格式:
参考:https://zhuanlan.zhihu.com/p/26506787
Illumina测序仪下机的数据通常为bcl格式,是将同一个测序通道(Lane)所有样品的数据混杂在一起。每个Lane里有n个样品的fastq.gz和一个undetermined.fastq.gz。区分每一样品的方法是根据不同的index。
Illumina官方出品的Bcl2FastQ软件,根据Index序列分割转换成每个样品的FastQ文件,打开长这样:
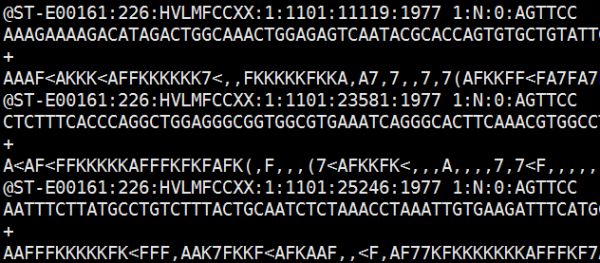
每一条read,包含四行,第一行read的ID,最后几个碱基序列是index;第二行是你的library里的DNA的序列(应该是不包括index和barcord primer 序列了);第三行+/-应该代表正链反链(具体没什么意义);第四行,每个碱基的测序质量。以上就是fastaq的嘴脸。
#常用软件#
我以前都是用cutadapt + FASTX-Toolkit的组合,直到同事们给我推荐了Trim Galore,质量评估使用FastQC。
BCL 格式文件是Illumina DNA sequencing instruments (HiSeq 或者 MiSeq) 创建的文件。BCL文件可以被CASAVA系统进行分析。也可以通过Illumina DNA sequencing instruments转化成QSEC格式文件。
Illumina Miseq的官方中文介绍
bcl文件的位置:
Miseq的Bcl文件位置在e.g.: /sequencedata/MiSeq/170808_M00528_0300_000000000-AP0TP/Data/Intensities/BaseCalls/L001/C1.1
我们的Miseq data是自动传入到服务器里的,我们连接到服务器后就可以进入到这个文件夹。
bcl2fastq2安装及其依赖gcc,boost,cmake等的安装
bcl2fastq2 Conversion v2.19 使用指导
bcl2fastq2 release NOTE 官网
bcl2fastq 软件常见的问题:
KNOWN ISSUES:
Corrupted *.bcl or *.bcl.gz files may cause bcl2fastq to stall indefinitely.
No index sequences are included in the header for each read in the resulting FASTQ
files if bcl2fastq is run without providing a sample sheet file.
The HTML report files will not display statistics for samples and projects named“default”, “all”, “unknown”, and “undetermined”.
The HTML report, Stats.json, and ConversionStats.xml files incorrectly reports the
% ≥ 30metric by excluding bases with quality score 30 (i.e. the number reported is
actually % > Q30).
5’ adapter trimming is not supported.
“N” is incorrectly allowed as anindex sequence character in the sample sheet. When
used, this will cause a mismatch for any sequence character other than “N”.
No warnings or errors are displayed when bcl2fastq is used to process run folders
that are missing control files.
Sample sheet files generated from Illumina Experiment Manager may cause bcl2fastq
to abort if they contain non-ASCII characters. Only alphanumeric characters dashes,
and underscores are allowed in the sample sheet.
在使用bcl2fastq时候sample sheet的正确格式:
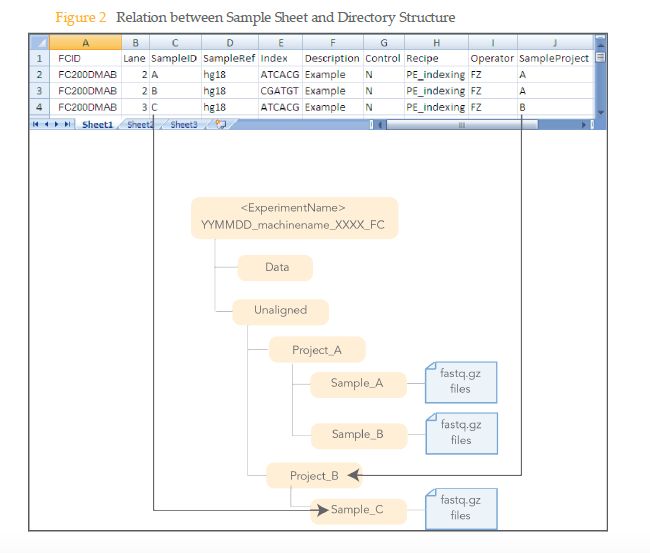
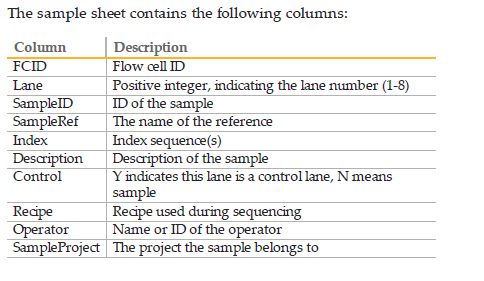
Illumina刚下机的数据为bcl格式文件(per-cycle BCL basecall file),但是下游的分析一般都需要fastq格式文件,所以在进行下游分析之前,需要使用CASAVA软件中的configureBclToFastq.pl将bcl格式的文件根据每个样本之前添加的index分出,并转为fastq格式的文件。在看bcl2fastq的说明文档时,会经常碰到一个词:demultiplexing,指的就是将multiplexed的reads根据index从不同或者同一个lane中分出,生成sample对应的fastq文件,这一步就涉及到输入正确的samplesheet.csv。
所有的步骤只使用一行代码就可以解决,首先贴出代码:

参考:chen_amiao的博客
以下参考:
bcl2fastq是illumina官方提供的bcl文件转化为fastq软件。
Google或官网搜索最新版,https://support.illumina.com/downloads/bcl2fastq-conversion-software-v217.html
下载
bcl2fastq2 Conversion Software v2.17 Installer (Linux tarball) 安装源文件
bcl2fastq2 Conversion Software v2.17 Guide (15051736 G) 介绍文件pdf
电脑Ubuntu14.04准备环境:
To build bcl2fastq2 Conversion Software v2.17, you need the following software.Versions listed are tested and supported; newer versions are untested.
} gcc 4.7 (with support for c++11)
} boost 1.54 (with its dependencies)
} CMake 2.8.9
} zlib
} librt
} libpthread
系统:bio-linux8
1.更新软件(安装环境)
sudo apt-get update
sudo apt-get upgrade
sudo apt-get install zlibc
sudo apt-get install libc6 # provides librt and libpthread
sudo apt-get install gcc
sudo apt-get install g++
sudo apt-get install libboost1.54-all-dev
sudo apt-get install cmake
#设置变量
export TMP=/tmpexport SOURCE=${TMP}/bcl2fastq
export BUILD=${TMP}/bcl2fastq2-v2.17.1.14-build
export INSTALL_DIR=/usr/local/bcl2fastq2-v2.17.1.14
cd ${TMP}
#软件包放在 /home/me/下载/bcl2fastq2/
tar -xvzf /home/me/下载/bcl2fastq2/bcl2fastq2-v2.17.1.14.tar.gz
mkdir ${BUILD}
cd ${BUILD}
sudo ${SOURCE}/src/configure --prefix=${INSTALL_DIR}
#上步显示成功,继续下面,未成功则可能是有些软件包没装好,重新更新下依赖环境
make
make install
#################测试##################
/usr/local/bcl2fastq2-v2.17.1.14/bin/bcl2fastq -v
2.运行参数 -h
/usr/local/bcl2fastq2-v2.17.1.14/bin/bcl2fastq -h
参考:http://nhoffman.github.io/borborygmi/compiling-bcl2fastq-on-ubuntu.html#sec-2
https://support.illumina.com/content/dam/illumina-support/documents/documentation/software_documentation/bcl2fastq/bcl2fastq2-v2-17-software-guide-15051736-g.pdf