教程对应B站:【生信技能树】生信人应该这样学R语言
配套资料:B站的11套生物信息学公益视频配套讲义、练习题及思维导图
先仔细观看视频,理解代码含义
中级题目
题目链接:http://www.bio-info-trainee.com/3750.html
- 探针ID转换
- 表达矩阵处理
- 任意基因任意癌症表达量和临床形状的关联
- 任意基因任意癌症表达量分组的生存分析
- 选取差异明显的基因的表达量绘制热图
- 表达矩阵样本的相关性
- 差异分析
想着为什么不能 TOC 呢?当然可以,教程在链接中。
左侧目录生成
探针ID转换
1、根据R包org.Hs.eg.db找到下面 ensembl 基因ID 对应的基因名(symbol) 关于此包的介绍:http://www.bio-info-trainee.com/710.html
ENSG00000000003.13
ENSG00000000005.5
ENSG00000000419.11
ENSG00000000457.12
ENSG00000000460.15
ENSG00000000938.11
rm(list = ls())
suppressMessages(library(org.Hs.eg.db))
keytypes(org.Hs.eg.db)
## [1] "ACCNUM" "ALIAS" "ENSEMBL" "ENSEMBLPROT"
## [5] "ENSEMBLTRANS" "ENTREZID" "ENZYME" "EVIDENCE"
## [9] "EVIDENCEALL" "GENENAME" "GO" "GOALL"
## [13] "IPI" "MAP" "OMIM" "ONTOLOGY"
## [17] "ONTOLOGYALL" "PATH" "PFAM" "PMID"
## [21] "PROSITE" "REFSEQ" "SYMBOL" "UCSCKG"
## [25] "UNIGENE" "UNIPROT"
ensembl_id <- c("ENSG00000000003.13","ENSG00000000005.5","ENSG00000000419.11","ENSG00000000457.12","ENSG00000000460.15","ENSG00000000938.11")
# 关于字符串处理包stringr介绍:https://stringr.tidyverse.org/
# 第一步:去掉 . 后的数字,得到ensembl_id
suppressMessages(library(stringr))
ensembl_id = str_split(ensembl_id, pattern = "[.]", simplify = T)[,1]
ensembl_id = as.data.frame(ensembl_id)
# 第二步:根据 org.Hs.eg.db 的作用获取对应关系
g2s = toTable(org.Hs.egSYMBOL)
g2e = toTable(org.Hs.egENSEMBL)
str(g2s)
## 'data.frame': 61119 obs. of 2 variables:
## $ gene_id: chr "1" "2" "3" "9" ...
## $ symbol : chr "A1BG" "A2M" "A2MP1" "NAT1" ...
g2e = toTable(org.Hs.egENSEMBL)
str(g2e)
## 'data.frame': 30903 obs. of 2 variables:
## $ gene_id : chr "1" "2" "3" "9" ...
## $ ensembl_id: chr "ENSG00000121410" "ENSG00000175899" "ENSG00000256069" "ENSG00000171428" ...
# 第三步:关联得到ensembl_id -> gene_id -> symbol的表格
# 根据ensembl_id在数据中找到对应的gene_id
ensembl = merge(ensembl_id, g2e, by='ensembl_id',all.x=T)
# 根据gene_id在数据库中找到对应的symbol(symbol是gene的名称)
gene = merge(ensembl, g2s, by = 'gene_id')
2、根据R包hgu133a.db找到下面探针对应的基因名(symbol)
平台对应的数据包网址:http://www.bio-info-trainee.com/1399.html
1053_at
117_at
121_at
1255_g_at
1316_at
1320_at
1405_i_at
1431_at
1438_at
1487_at
1494_f_at
1598_g_at
160020_at
1729_at
177_at
下载安装这个包:https://bioconductor.org/packages/release/data/annotation/html/hgu133a.db.html
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("hgu133a.db", version = "3.8")
suppressMessages(library(hgu133a.db))
# 读入探针id文件,列名改为probe_id
probe_id = read.table(file='probe_id.txt',head=F)
colnames(probe_id) = 'probe_id'
ids = toTable(hgu133aSYMBOL)
gene_p = merge(probe_id,ids,by='probe_id',all.x=T)
rm(list = ls())
表达矩阵处理
表达矩阵分析大全可以学习GEO数据挖掘:https://www.bilibili.com/video/av26731585/
3、找到R包CLL内置的数据集的表达矩阵里面的TP53基因的表达量,并且绘制在 progres.-stable 分组的boxplot图
# 下载安装这个包:http://bioconductor.org/packages/release/data/experiment/html/CLL.html
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("CLL", version = "3.8")
suppressPackageStartupMessages(library(CLL))
# http://bioconductor.org/packages/release/data/experiment/manuals/CLL/man/CLL.pdf
data(sCLLex)
# exprs()函数提取表达矩阵
exprSet = exprs(sCLLex)
# pData()函数查看该对象的样本分组信息
pd = pData(sCLLex)
str(pd)
## 'data.frame': 22 obs. of 2 variables:
## $ SampleID: 'AsIs' chr "CLL11" "CLL12" "CLL13" "CLL14" ...
## $ Disease : Factor w/ 2 levels "progres.","stable": 1 2 1 1 1 1 2 2 1 2 ...
# 加载注释包
suppressPackageStartupMessages(library(hgu95av2.db))
ids = toTable(hgu95av2SYMBOL)
# 在ids中搜索TP53得到三个探针
# a %in% table 判断a值是否在table中
probe <- ids[ids$symbol%in%"TP53",][,1]
boxplot(exprSet["1939_at",] ~ pd$Disease)
boxplot(exprSet['1974_s_at',] ~ pd$Disease)
boxplot(exprSet['31618_at',] ~ pd$Disease)
#ggpubr
d <-cbind(as.data.frame(exprSet['1939_at',]),as.data.frame(pd$Disease))
head(d)
suppressPackageStartupMessages(library(ggpubr))
colnames(d)<-c("e","Disease")
p<-ggboxplot(d, x="Disease", y ="e",
palette = "jco",
add = "jitter")
# 设置图形的参数
opar<-par(no.readonly=T)
# Add p-value
p + stat_compare_means()
# Change method
p + stat_compare_means(method = "t.test")
# 其余图片未展示,展示最后一张:
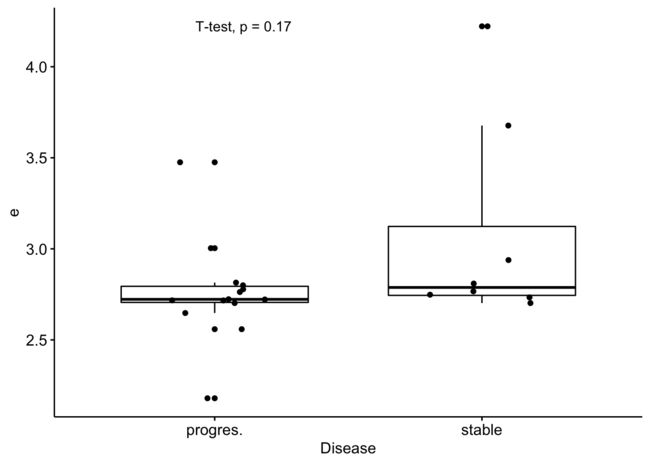
任意基因任意癌症表达量和临床形状的关联
4、找到BRCA1基因在TCGA数据库的乳腺癌数据集(Breast Invasive Carcinoma (TCGA, PanCancer Atlas))的表达情况
cbioportal地址:http://www.cbioportal.org/
cgdsR教程:http://www.bio-info-trainee.com/1257.html
BRCA1 <- read.table('plot-BRCA1.txt',sep='\t',fill=T,header = T)
colnames(BRCA1) = c('id','subtype','expression','mutant')
suppressPackageStartupMessages(library(ggstatsplot))
ggbetweenstats(data = BRCA1, x="subtype", y="expression")
suppressPackageStartupMessages(library(ggpubr))
ggboxplot(data=BRCA1, x="subtype", y="expression", color = "subtype",add = "point", shape="subtype")
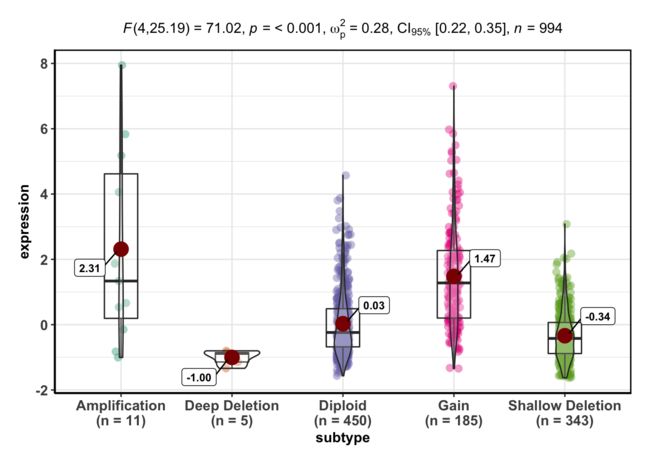
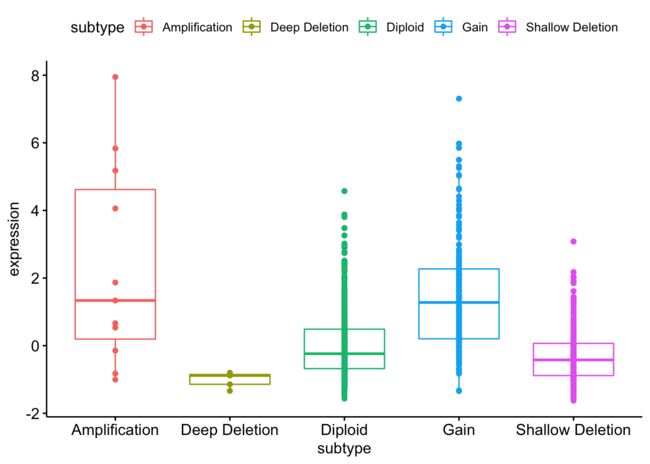
任意基因任意癌症表达量分组的生存分析
5、找到TP53基因在TCGA数据库的乳腺癌数据集的表达量分组看其是否影响生存
提示使用:http://www.oncolnc.org/
TCGA数据库生存分析的网页工具哪家强:http://www.bio-info-trainee.com/3783.html
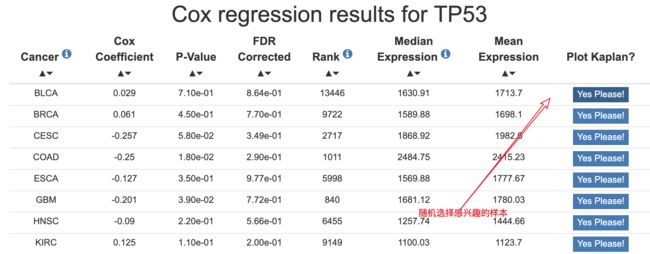
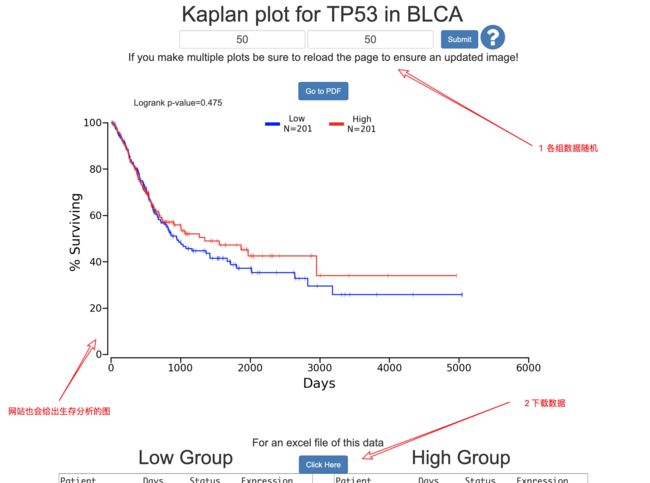
#随机选择了BLCA的TP53,50 50 分为高表达和低表达组
BLCA = read.csv(file='BLCA_7157_50_50.csv',header = T)
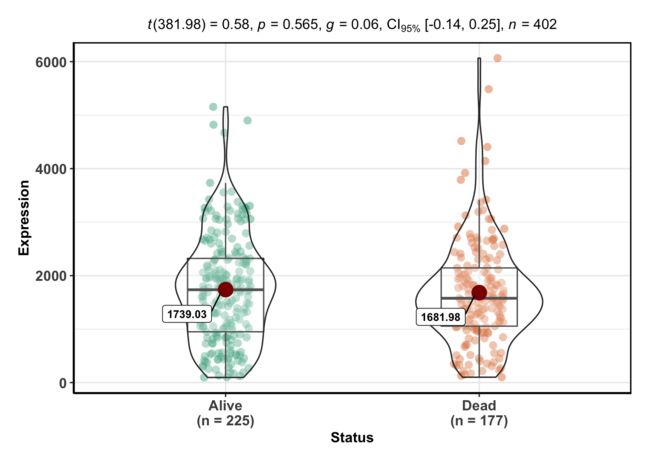
ggbetweenstats(data = BLCA, x='Status', y='Expression')
suppressPackageStartupMessages(library(survival))
suppressPackageStartupMessages(library(survminer))
table(BLCA$Status)
## Alive Dead
## 225 177
BLCA$Status = ifelse(BLCA$Status=='Dead',1,0)
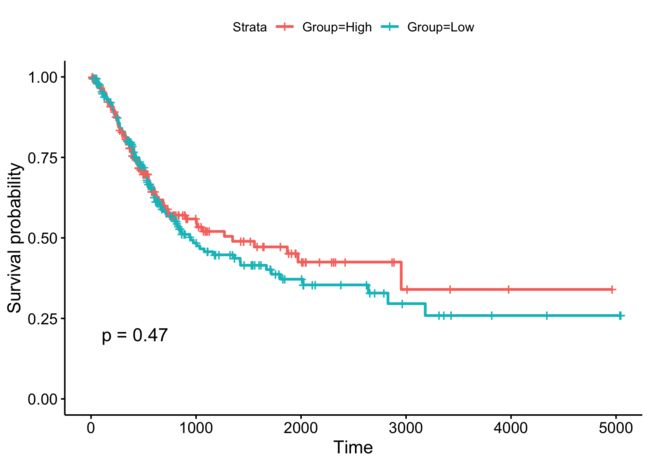
sfit <- survfit(Surv(Days,Status)~Group,data = BLCA)
ggsurvplot(sfit, conf.int = F, pval=T)
rm(list = ls())
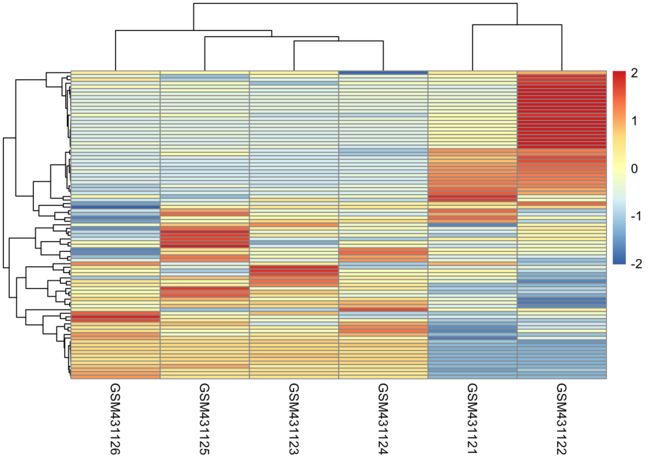
选取差异明显的基因的表达量绘制热图
6、下载数据集GSE17215的表达矩阵并且提取下面的基因画热图
ACTR3B ANLN BAG1 BCL2 BIRC5 BLVRA CCNB1 CCNE1 CDC20 CDC6 CDCA1 CDH3 CENPF CEP55 CXXC5 EGFR ERBB2 ESR1 EXO1 FGFR4 FOXA1 FOXC1 GPR160 GRB7 KIF2C KNTC2 KRT14 KRT17 KRT5 MAPT MDM2 MELK MIA MKI67 MLPH MMP11 MYBL2 MYC NAT1 ORC6L PGR PHGDH PTTG1 RRM2 SFRP1 SLC39A6 TMEM45B TYMS UBE2C UBE2T
# 关于 GEOquery 下载数据 :http://www.bio-info-trainee.com/941.html
# GEOquery 的文档:https://www.rdocumentation.org/packages/GEOquery/versions/2.38.4
suppressPackageStartupMessages(library(GEOquery))
gse17215 = getGEO("GSE17215", destdir=".", AnnotGPL = F, getGPL = F)
gse17215 = gse17215$GSE17215_series_matrix.txt.gz
gse17215 = as.data.frame(exprs(gse17215))
dim(gse17215)
## [1] 22277 6
suppressPackageStartupMessages(library(hgu133a.db))
ids = toTable(hgu133aSYMBOL)
# 将基因写入txt,读取
gene <- read.table("GSE17215-gene.txt",sep=" ",header = F)
gene = as.data.frame(t(gene),row.names=F)
colnames(gene) = "symbol"
symbol = merge(gene,ids,by="symbol")
gse17215$probe_id = rownames(gse17215)
probe_id = merge(symbol,gse17215,by="probe_id")
rownames(probe_id) = probe_id[,1]
probe_id = probe_id[,c(-1,-2)]
suppressPackageStartupMessages(library(pheatmap))
n = t(scale(t(probe_id))) # scale()函数去中心化和标准化
# 对每个探针的表达量进行去中心化和标准化
n[1:4,1:4]
## GSM431121 GSM431122 GSM431123 GSM431124
## 201397_at -0.2574111 -1.8106578 0.7780733 0.93838912
## 201710_at -0.4562536 0.3155208 0.3556447 -0.07541007
## 201820_at -1.3293345 -1.2475826 0.7165131 0.56294891
## 201890_at 1.0811204 1.4793394 -0.6189666 -0.60361912
pheatmap(n,show_rownames = F,clustering_distance_rows = "correlation")
rm(list = ls())
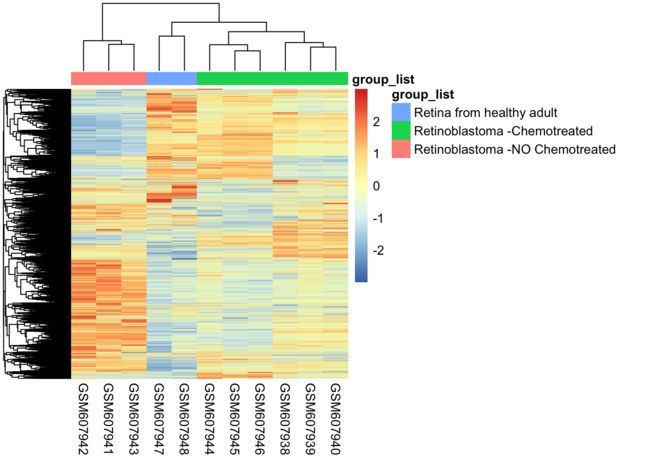
表达矩阵样本的相关性
7、下载数据集GSE24673的表达矩阵计算样本的相关性并且绘制热图,需要标记上样本分组信息
suppressPackageStartupMessages(library(GEOquery))
gse24673 = getGEO("GSE24673",destdir=".", AnnotGPL = F, getGPL = F)
gse24673 = gse24673$GSE24673_series_matrix.txt.gz
# 得到表达矩阵和样本分组
gse24673_f = as.data.frame(exprs(gse24673))
gse24673_pd = pData(gse24673)
gse24673_group = gse24673_pd[,"source_name_ch1"]
gse24673_group = as.data.frame(gse24673_group,row.names = rownames(gse24673_pd))
colnames(gse24673_group) = "group_list"
# 绘图
pheatmap(gse24673_f,scale = "row",show_rownames = F, annotation_col = gse24673_group)
rm(list = ls())
8、找到 GPL6244 platform of Affymetrix Human Gene 1.0 ST Array 对应的R的bioconductor注释包,并且安装它!
# 文章;https://www.jianshu.com/p/f6906ba703a0
BiocManager::install("hugene10sttranscriptcluster.db")
9、下载数据集GSE42872的表达矩阵,并且分别挑选出 所有样本的(平均表达量/sd/mad/)最大的探针,并且找到它们对应的基因。
suppressPackageStartupMessages(library(GEOquery))
gse42872 = getGEO("GSE42872", destdir=".", AnnotGPL = F, getGPL = F)
gse42872 = gse42872$GSE42872_series_matrix.txt.gz
gse42872_f = as.data.frame(exprs(gse42872))
sort(apply(gse42872_f,1,mean),decreasing = T)[1]
## 7978905
## 14.53288
sort(apply(gse42872_f,1,sd),decreasing = T)[1]
## 8133876
## 3.166429
sort(apply(gse42872_f,1,mad),decreasing = T)[1]
## 8133876
## 4.268561
suppressPackageStartupMessages(library("hugene10sttranscriptcluster.db"))
ids = toTable(hugene10sttranscriptclusterSYMBOL)
mean = ids[ids$probe_id%in%7978905,]
sd = ids[ids$probe_id%in%8133876,]
差异分析
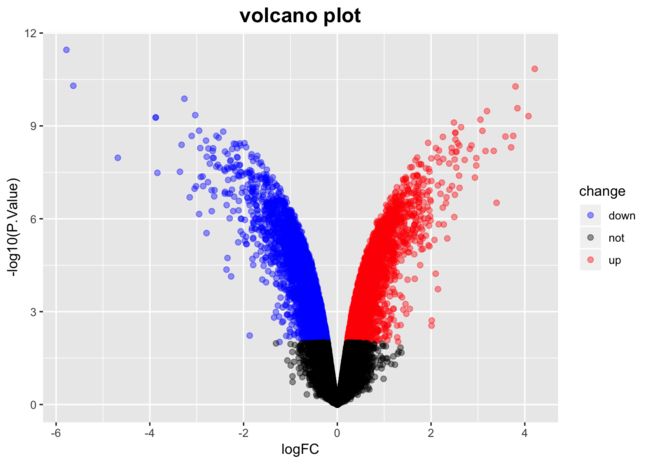
10、下载数据集GSE42872的表达矩阵,并且根据分组使用limma做差异分析,得到差异结果矩阵
limma 做差异分析:http://www.bio-info-trainee.com/bioconductor_China/software/limma.html
gse42872_pd = pData(gse42872)
gse42872_group = gse42872_pd[,"source_name_ch1"]
gse42872_group = as.data.frame(gse42872_group, row.names = rownames(gse42872_pd))
colnames(gse42872_group) = "group_list"
suppressPackageStartupMessages(library(stringr))
gse42872_group_list = as.data.frame(str_split(gse42872_group$group_list,pattern = " ", simplify = T)[,6],row.names = rownames(gse42872_group))
colnames(gse42872_group_list) = "group_list"
suppressMessages(library(limma))
design <- model.matrix(~0+factor(gse42872_group_list$group_list))
colnames(design)=c("vechile","vemurafenib")
rownames(design)=rownames(gse42872_group_list)
design
## vechile vemurafenib
## GSM1052615 1 0
## GSM1052616 1 0
## GSM1052617 1 0
## GSM1052618 0 1
## GSM1052619 0 1
## GSM1052620 0 1
## attr(,"assign")
## [1] 1 1
## attr(,"contrasts")
## attr(,"contrasts")$`factor(gse42872_group_list$group_list)`
## [1] "contr.treatment"
contrast<-makeContrasts("vechile-vemurafenib",levels = design)
contrast
DEG <- function(gse42872_f,design,contrast){
fit = lmFit(gse42872_f, design)
fit2 = contrasts.fit(fit, contrast)
fit2 = eBayes(fit2)
mtx = topTable(fit2, coef=1, n=Inf)
deg_mtx = na.omit(mtx)
return(deg_mtx)
}
DEG_mtx = DEG(gse42872_f,design,contrast)
plot(DEG_mtx$logFC, -log10(DEG_mtx$P.Value))
suppressPackageStartupMessages(library(ggplot2))
logFC_cutof = with(DEG_mtx,mean(abs(logFC)) + 2*sd(abs(logFC)))
logFC_cutof = 0
DEG_mtx$change = as.factor(ifelse(DEG_mtx$P.Value<0.01 & abs(DEG_mtx$logFC)>logFC_cutof,
ifelse(DEG_mtx$logFC>logFC_cutof,'up','down'),'not'))
ggplot(data = DEG_mtx, aes(x = logFC, y = -log10(P.Value), color = change)) +
geom_point(alpha = 0.4, size = 1.75) +
scale_color_manual(values=c("blue", "black","red"))+
labs(title = "volcano plot")+
theme(plot.title = element_text(size = 16, hjust = 0.5, face = "bold"))
更多学习资源:
生信技能树公益视频合辑
生信技能树账号
生信工程师入门最佳指南
...
欢迎关注生信菜鸟团、生信技能树!