送书 | 哈佛大学单细胞课程:笔记汇总前篇
经典赏析
NGS系列文章包括NGS基础、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述))、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step) - Limma差异分析、火山图、功能富集)等内容。
单细胞转录组测序进展迅速,伴随而来的是许许多多的内容与讲义,很多课程都老长。。。嗨像我这样莫有耐心的小孩子,能完整看完的只有一个 —— Single-cell RNA-seq analysis workshop(https://github.com/hbctraining/scRNA-seq)。我将把教程内容尽量汇总,你需要做的就是点击收藏慢慢看!
(一)Why single-cell RNA-seq
在整个人体组织中,细胞类型、状态和相互作用非常多样。为了更好地了解这些组织存在的细胞类型,单细胞RNA-seq(scRNA-seq)提供了在单细胞水平上表达基因的信息。
单细胞转录组测序可用于 :
探索组织中存在哪些细胞类型
识别未知/稀有的细胞类型或状态
阐明分化过程中或跨时间或状态的基因表达变化
识别在不同条件(例如治疗或疾病)下特定细胞类型中差异表达的基因
探索细胞类型之间的表达变化,同时纳入空间、调控和/或蛋白质信息
常见方法包括:

Challenges of scRNA-seq analysis
在scRNA-seq之前,我们通常使用bulk RNA-seq进行转录组分析。bulk RNA-seq是一种直接比较细胞的平均表达值的方法,在寻找疾病生物标志物,或者不关心样品中大量细胞异质性的情况下,这可能是最佳方法。
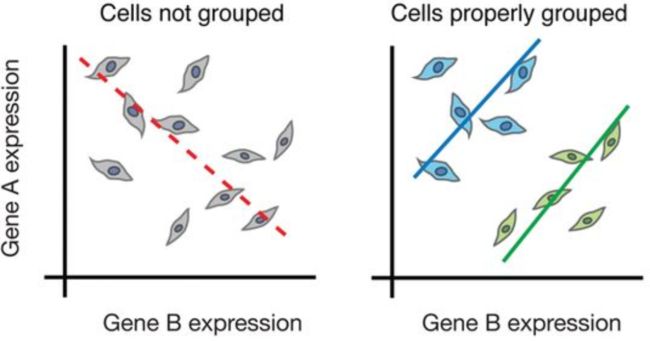
尽管bulk RNA-seq可以探索不同条件(例如治疗或疾病)之间基因表达的差异,但无法充分捕获细胞水平的差异。例如,在下面的图像中,如果进行bulk分析(左),我们将无法检测到基因A和基因B的表达之间的正确关联。但是,如果我们按细胞类型或细胞状态正确地对细胞进行分组,我们可以看到基因之间的正确相关性。
scRNA-seq也有一定的局限性,除了制样和建库价格高昂外,它在数据分析中也具有一定的复杂性,包括:
数据量大
细胞的测序深度低
细胞/样品之间的技术差异
跨细胞/样品的生物变异性
数据量大
scRNA-seq实验的数据来自捕获的成千上万个细胞,也就是数十万条reads,需要更多的内存和存储空间。
细胞的测序深度低
基于液滴的scRNA-seq方法的测序深度较浅,通常每个细胞只能检测到转录组的10-50%。这导致细胞中许多基因的计数为零。但是,在特定的细胞中,基因的零计数可能意味着该基因未表达或仅表示该转录本未被检测到。在整个细胞中,具有较高表达水平的基因测到0值的机率较低。由于此特征,在任何细胞中都不会检测到许多基因,并且细胞之间的基因表达差异很大。
Zero-inflated?:scRNA-seq data is often referred to as zero-inflated; however, recent analyses suggest that it does not contain more zeros than what would be expected given the sequencing depth (Valentine Svensson’s blog post:http://www.nxn.se/valent/2017/11/16/droplet-scrna-seq-is-not-zero-inflated).
跨细胞/样品的生物变异性
我们不感兴趣的某些生物变异可能导致细胞之间的基因表达比实际生物细胞的类型/状态更为相似/不同,并掩盖细胞类型。这些变异(除非实验研究的一部分)包括(以下为原文):
Transcriptional bursting: Gene transcription is not turned on all of the time for all genes. Time of harvest will determine whether gene is on or off in each cell.
Varying rates of RNA processing: Different RNAs are processed at different rates.
Continuous or discrete cell identities (e.g. the pro-inflammatory potential of each individual T cell): Continuous phenotypes are by definitition variable in gene expression, and separating the continuous from the discrete can sometimes be difficult.
Environmental stimuli: The local environment of the cell can influence the gene expression depending on spatial position, signaling molecules, etc.
Temporal changes: Fundamental fluxuating cellular processes, such as cell cycle, can affect the gene expression profiles of individual cells.

Image credit: Wagner, A, et al. Revealing the vectors of cellular identity with single-cell genomics, Nat Biotechnol. 2016 (doi:https://dx.doi.org/10.1038%2Fnbt.3711)
细胞/样品之间的技术差异
细胞特异性捕获效率:不同细胞捕获的转录本不同,导致测序深度不同(例如,转录组的10-50%)。
文库质量:降解的RNA、低存活力/濒死细胞、大量自由漂浮的RNA以及细胞定量不准确会导致质量指标降低。
扩增偏差:在文库制备的扩增步骤中,并非所有转录本都扩增到相同水平。
批次效应:对于scRNA-Seq分析,批次效应是一个重要的问题,因为看到的显著差异表达可能只是因为批次效应引起的。

Image credit: Hicks SC, et al., bioRxiv (2015)(https://www.biorxiv.org/content/early/2015/08/25/025528)
批次不良所产生的问题在这篇文章中有很好的介绍:https://f1000research.com/articles/4-121/v1
如何知道你的实验中具有批次呢?
是否在同一天进行了所有RNA的分离?
是否在同一天进行了所有建库工作?
是否由同一个人对所有样品进行RNA分离/文库制备?
是否对所有样品使用相同的试剂?
是否在同一位置进行RNA分离/文库制备?
如果以上任一问题的答案为“否”,说明你的实验中具有批次。
有关批次的最佳做法:
尽可能避免以批次的方式设计实验。
如果无法避免:
Do NOT confound your experiment by batch:

将不同样品组的重复样品分成多个批次:

在实验设计文件中加上批次信息,这样可以在分析过程中退还批次引起的差异。
建议:
在实验开始之前与专家讨论实验设计。
同时从样品中分离RNA。
同时准备样品库或备用样品组,以避免批次混淆。
不要混淆性别、年龄或批次的样本组。
(二)Single-cell RNA-seq data - raw data to count matrix
根据所用文库制备方法的不同,RNA序列(也被称为reads或tag)将从转录本((10X Genomics, CEL-seq2, Drop-seq, inDrops)的3'端(或5'端)或全长转录本(Smart-seq)中获得。

Image credit: Papalexi E and Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity, Nature Reviews Immunology 2018 (https://doi.org/10.1038/nri.2017.76)
不同测序方式的优点:
3’(或5’)末端测序:
通过使用UMI进行更准确的定量,从而将生物学重复与扩增重复(PCR)区别开来;
测序的细胞数量更多,可以更好地鉴定细胞类型群;
每个细胞成本更低;
大于10,000个细胞的结果最佳
全长测序:
检测亚型水平(
isoform-level)表达差异;鉴定等位基因特异性差异表达;
对较少数量的细胞进行更深的测序;
最适用于细胞数少的样品。
我们将主要介绍3’端测序,重点是基于液滴的方法(inDrops, Drop-seq, 10X Genomics)。
3’-end reads (includes all droplet-based methods)
在3’端测序中,同一转录本的不同reads片段仅会源自转录本的3’端,相同序列的可能性很高,同时在建库过程中的PCR步骤可能导致reads的重复,因此为了区分是生物学还是技术上的重复,我们使用唯一标识符(unique molecular identifiers,UMI)进行标注。
比对到相同的转录本、UMI不同的reads来源于不同的分子,为生物学重复,每个read都被计数。
UMI相同的reads来自同一分子,为技术重复,计为1个read。
我们以下图为例,下图中分子ACTB的UMI均相同,因此只能记为1个molecule,而ARL1的UMI不同所以可以记为2个molecule。

Image credit: modified from Macosko EZ et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets, Cell 2015 (https://doi.org/10.1016/j.cell.2015.05.002)
在细胞水平进行正确定量都需要以下条件:
Sample index: 样本来源
Added during library preparation - needs to be documented
Cellular barcode: 细胞来源
Each library preparation method has a stock of cellular barcodes used during the library preparation
Unique molecular identifier (UMI): 转录本来源
The UMI will be used to collapse PCR duplicates
Sequencing read1: the Read1 sequence
Sequencing read2: the Read2 sequence
例如,使用inDrops v3库准备方法时,以下内容是reads的所有信息:

Image credit: Sarah Boswell(https://scholar.harvard.edu/saboswell), Director of the Single Cell Sequencing Core at HMS
R1 (61 bp Read 1): sequence of the read (Red top arrow)
R2 (8 bp Index Read 1 (i7)): cellular barcode - which cell read originated from (Purple top arrow)
R3 (8 bp Index Read 2 (i5)): sample/library index - which sample read originated from (Red bottom arrow)
R4 (14 bp Read 2): read 2 and remaining cellular barcode and UMI - which transcript read originated from (Purple bottom arrow)
对于不同的基于液滴的scRNA-seq方法,scRNA-seq的分析工作流程相似,但是UMI、细胞ID和样品索引的解析会有所不同。例如,以下是10X序列reads的示意图,其中index,UMI和barcode的位置不同:
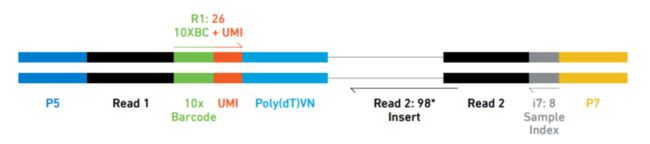
Image credit: Sarah Boswell(https://scholar.harvard.edu/saboswell), Director of the Single Cell Sequencing Core at HMS
Single-cell RNA-seq workflow
scRNA-seq方法能通过测序的reads解析barcodes和UMI,它们在特定步骤里会轻微地不同,但除了方法外,大致流程都是一致的,常规工作流程如下所示:

Image credit: Luecken, MD and Theis, FJ. Current best practices in single‐cell RNA‐seq analysis: a tutorial, Mol Syst Biol 2019 (doi: https://doi.org/10.15252/msb.20188746)
重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程 (原理、代码和评述)
工作流程的步骤是:
生成count矩阵(
method-specific steps):reads格式化,对样本进行多路分解(demultiplexing,即通过barcodes确定reads的来源),比对和定量。原始count的质量控制:过滤质量较差的细胞。
过滤count的聚类:基于转录活性的相似性对细胞进行聚类(细胞类型数=簇数)。
marker识别:识别每个cluster的标记基因。可选的下游步骤。
无论进行那种分析,生物学重复都是必要的!
Generation of count matrix
我们聚焦于基于液滴型的3’端测序(比如inDrops、10X Genomics和Drop-seq),将原始测序数据转换为count矩阵。
测序工具将以BCL或FASTQ格式输出原始测序数据,或生成count矩阵。如果reads是BCL格式,我们将需要转换为FASTQ格式。有一个有用的命令行工具bcl2fastq,可以轻松执行此转换。
NOTE: We do not demultiplex at this step in the workflow. You may have sequenced 6 samples, but the reads for all samples may be present all in the same BCL or FASTQ file.
对于许多scRNA-seq方法,从原始测序数据中生成count矩阵都将经历相似的步骤。

umis(https://github.com/vals/umis)和zUMIs(https://github.com/vals/umis)是命令行工具,可用于估计测转录本3'端的scRNA-seq数据的表达。此过程中的步骤包括:
格式化reads并过滤嘈杂的细胞
barcodes;Demultiplexing the samples(通过barcodes确定reads的来源);比对/伪比对到转录;
折叠UMI和定量reads。
当然,如果使用10X Genomics建库方法,Cell Ranger pipeline(https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger)将负责执行以上的所有步骤。
格式化reads并过滤嘈杂的细胞barcodes:
FASTQ文件能解析得到细胞barcodes、UMIs和样本barcodes。对于基于液滴型的方法,一些细胞barcodes会对应的低的reads数(< 1000 reads) ,原因是:
encapsulation of free floating RNA from dying cells
simple cells (RBCs, etc.) expressing few genes
cells that failed for some reason
在比对reads之前,需要从序列数据中过滤掉多余的条形码。为了进行这种过滤,提取并保存每个细胞的“细胞条形码”和“分子条形码”。例如,如果使用“umis”工具,则信息将以以下格式添加到每次reads的标题行中:
@HWI-ST808:130:H0B8YADXX:1:1101:2088:2222:CELL_GGTCCA:UMI_CCCT
AGGAAGATGGAGGAGAGAAGGCGGTGAAAGAGACCTGTAAAAAGCCACCGN
+
@@@DDBD>=AFCF+建库中使用的细胞条形码应该是已知的,未知的条形码会被丢弃,同时对于已知的细胞条形码可以有一定量的错配。
Demultiplexing the samples:
如果测序多于一个样品执行此步骤,这是一步不由“umis”工具处理,而由“zUMIs”完成的步骤,这步会解析reads以确定与每个与细胞相关的样本条形码。
比对/伪比对到转录:
通过传统(STAR)或轻量型(Kallisto/RapMap)方法,将reads比对回基因。
折叠UMI和定量reads:
使用Kallisto或featureCounts之类的工具仅对唯一的UMI进行量化,得到

Image credit: extracted from Lafzi et al. Tutorial: guidelines for the experimental design of single-cell RNA sequencing studies, Nature Protocols 2018 (https://doi.org/10.1038/s41596-018-0073-y)
矩阵中的每个值代表源自相应基因的细胞中的reads数。
(三)Single-cell RNA-seq: Quality control set-up

在生成count矩阵后,我们需要对其进行QC分析,并将其导入R进行后续步骤:
探索示例集
该数据集来自Kang et al, 2017(https://www.nature.com/articles/nbt.4042)的文章,是八名狼疮患者的PBMC(Peripheral Blood Mononuclear Cells)数据,将其分为对照组和干扰素beta处理(刺激)组。

Image credit: Kang et al, 2017(https://www.nature.com/articles/nbt.4042)
Raw data
研究团队发现GEO (GSE96583:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96583)提供的矩阵缺少线粒体的reads,因此从SRA (SRP102802:https://www-ncbi-nlm-nih-gov.ezp-prod1.hul.harvard.edu/sra?term=SRP102802)下载BAM文件,然后转化为FASTQ文件,并使用Cell Ranger(https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger)获得count矩阵。
NOTE: The counts for this dataset is also freely available from 10X Genomics and is used as part of the
Seurat tutorial(https://satijalab.org/seurat/v3.0/immune_alignment.html).(我说我咋这么眼熟。。。)
Metadata
相关信息也叫metadata。关于以上数据的metadata如下:
使用10X Genomics版本2化学试剂制备文库;
样品在Illumina NextSeq 500上测序;
将来自八名狼疮患者的PBMC样本分别分成两等份:
一份用100 U/mL重组IFN-β激活PBMC,处理时间为6小时。
另一份样品未经处理。
6小时后,将不同条件的8个样品一起收集起来(受激细胞和对照细胞)。
分别鉴定了
12138和12167个细胞(去除doublets后)作为对照样品和刺激后的合并样品。由于样品是PBMC,因此我们期望有免疫细胞,例如:
B cells
T cells
NK cells
monocytes
macrophages
possibly megakaryocytes
It is recommended that you have some expectation regarding the cell types you expect to see in a dataset prior to performing the QC. This will inform you if you have any cell types with low complexity (lots of transcripts from a few genes) or cells with higher levels of mitochondrial expression. This will enable us to account for these biological factors during the analysis workflow.
上述细胞类型都不预估有低复杂度或高线粒体含量。
设置R环境
为了更好的管理数据,使得整个项目具有组织性,先创建一个项目叫“single_cell_rnaseq”,然后构建以下目录:
single_cell_rnaseq/
├── data
├── results
└── figures下载数据
Control sample(https://www.dropbox.com/sh/73drh0ipmzfcrb3/AADMlKXCr5QGoaQN13-GeKSa?dl=1)
Stimulated sample(https://www.dropbox.com/sh/cii4j356moc08w5/AAC2c3jfvh2hHWPmEaVsZKRva?dl=1)
解压数据后新建一个R脚本,命名为“quality_control.R”,并且保证整个的工作目录看起来像这样:

加载R包
library(SingleCellExperiment)
library(Seurat)
library(tidyverse)
library(Matrix)
library(scales)
library(cowplot)
library(RCurl)加载count矩阵
一共有3个文件,分别为:分别为cell IDs、gene IDs和matrix of counts(每个细胞的每个基因)。
barcodes.tsv 这是一个文本文件,其中包含该样品存在的所有细胞条形码。条形码以矩阵文件中显示的数据顺序列出(这些是列名)。
这是一个文本文件,其中包含该样品存在的所有细胞条形码。条形码以矩阵文件中显示的数据顺序列出(这些是列名)。features.tsv 这是一个文本文件,其中包含定量基因的标识符。标识符的来源可能会有所不同,具体取决于定量过程中使用的参考(即
这是一个文本文件,其中包含定量基因的标识符。标识符的来源可能会有所不同,具体取决于定量过程中使用的参考(即Ensembl,NCBI,UCSC),大多数情况下这些都是官方gene symbol。这些基因的顺序与矩阵文件中各行的顺序相对应(这些是行名)。matrix.mtx
注意该矩阵中有许多zero values。
有两种方法可以进行矩阵读入,分别为readMM()和Read10X()。
readMM(): This function is from the Matrix package and will turn our standard matrix into a sparse matrix. The features.tsv file and barcodes.tsv must first be individually loaded into R and then they are combined. For specific code and instructions on how to do this please see our additional material(https://github.com/hbctraining/scRNA-seq/blob/master/lessons/readMM_loadData.md).
Read10X(): This function is from the Seurat package and will use the Cell Ranger output directory as input. In this way individual files do not need to be loaded in, instead the function will load and combine them into a sparse matrix for you. We will be using this function to load in our data!
Reading in a single sample (read10X())
我们通常使用Read10X()。原因就要从头说起了,一般在软件Cell Ranger处理10X数据后会生成一个outs目录,在outs文件夹中有这样几个文件:
web_summary.html: 包括许多QC指标,预估细胞数,比对率等;
BAM alignment files:用于可视化比对的reads和重新创建FASTQ文件;
filtered_feature_bc_matrix:经过Cell Ranger过滤后构建矩阵所需要的所有文件;
raw_feature_bc_matrix: 未过滤的可以用于构建矩阵的文件;
如果你有一个单个样本的话,可以直接构建a Seurat object(https://github.com/satijalab/seurat/wiki/Seurat):
# How to read in 10X data for a single sample (output is a sparse matrix)
ctrl_counts <- Read10X(data.dir = "data/ctrl_raw_feature_bc_matrix")
# Turn count matrix into a Seurat object (output is a Seurat object)
ctrl <- CreateSeuratObject(counts = ctrl_counts,
min.features = 100)NOTE: The min.features argument specifies the minimum number of genes that need to be detected per cell. This argument will filter out poor quality cells that likely just have random barcodes encapsulated without any cell present. Usually, cells with less than 100 genes detected are not considered for analysis.
在使用Read10X()函数读取数据时,Seurat会为每个细胞自动创建metadata,存储在meta.dataslot。
The Seurat object is a custom list-like object that has well-defined spaces to store specific information/data. You can find more information about the slots in the Seurat object at this link:https://github.com/satijalab/seurat/wiki/Seurat
# Explore the metadata
head([email protected])metadata 的每一列代表什么呢?
orig.ident:细胞聚类的cluster,含有样本的身份信息(已知的情况下);nCount_RNA:每个细胞的UMI数目;nFeature_RNA:检测到的每个细胞中的genes数目。
使用for循环对多个数据进行读入
# Create each individual Seurat object for every sample
for (file in c("ctrl_raw_feature_bc_matrix", "stim_raw_feature_bc_matrix")){
seurat_data <- Read10X(data.dir = paste0("data/", file))
seurat_obj <- CreateSeuratObject(counts = seurat_data,
min.features = 100,
project = file)
assign(file, seurat_obj)
}于是分别形成了ctrl_raw_feature_bc_matrix和stim_raw_feature_bc_matrix两个seurat对象,使用merge函数对不同数据进行合并:
# Create a merged Seurat object
merged_seurat <- merge(x = ctrl_raw_feature_bc_matrix,
y = stim_raw_feature_bc_matrix,
add.cell.id = c("ctrl", "stim"))因为相同的细胞ID可以用于不同的样本,所以我们使用add.cell.id参数为每个细胞ID添加特定于样本的前缀。如果我们查看合并对象的metadata,我们应该能够在行名中看到前缀:
# Check that the merged object has the appropriate sample-specific prefixes
head([email protected])
tail([email protected])(四)Single-cell RNA-seq: Quality control
在QC步骤中,我们的目标和挑战主要包括:
目标:
筛选数据以仅包括高质量的真实细胞,以便在对细胞进行聚类时,更容易识别不同的细胞类型.
识别任何失败的样本并尝试挽救数据或从分析中删除,此外还试图了解样本失败的原因.
挑战:
从不太复杂的细胞中划定质量较差的细胞。
选择合适的阈值进行过滤,保留高质量的细胞而不去除生物学相关的细胞类型。
建议:
在执行质量控制之前,需要对细胞类型进行一个预估。例如,您是否需要样品中的复杂性较低的细胞或线粒体表达水平较高的细胞?如果是,那么在评估我们的数据质量时,我们需要考虑这种生物学特征。
生成质控指标:
除了我们上面提到过的orig.ident、nCount_RNA和nFeature_RNA外,我们还可以计算其他QC指标如number of genes detected per UMI(能反映数据的复杂度,UMI数越大数据复杂度越高)和mitochondrial ratio(可以得知来源于线粒体基因的reads)。
#将每个细胞的UMI数量进行log10转换并加入到metadata
merged_seurat$log10GenesPerUMI <- log10(merged_seurat$nFeature_RNA)/log10(merged_seurat$nCount_RNA)#计算线粒体相关基因比例,注意!("^MT-")只限于人哦。。。merged_seurat$mitoRatio <- PercentageFeatureSet(object = merged_seurat, pattern = "^MT-")
merged_seurat$mitoRatio <- [email protected]$mitoRatio / 100当然,我们也可以使用 $ 添加其他质控指标到metadata中(R变量索引 - 什么时候使用 @或$),这样不会影响merged_seurat对象。我们可以直接从Seurat对象中提取meta.dataslot 创建metadata:
# Create metadata dataframe
metadata <- [email protected]你应该看到每个细胞ID都有一个ctrl_或stim_前缀,这是我们合并Seurat对象时指定的,它们应与orig.ident中列出的样本匹配。我们可以先添加带有细胞ID的列,并更改当前列名称以使其更加直观:
# Add cell IDs to metadata
metadata$cells <- rownames(metadata)
# Rename columns
metadata <- metadata %>%
dplyr::rename(seq_folder = orig.ident,
nUMI = nCount_RNA,
nGene = nFeature_RNA)基于细胞前缀得到每个细胞的样本名。给sample这一列进行命名:
# Create sample column
metadata$sample <- NA
metadata$sample[which(str_detect(metadata$cells, "^ctrl_"))] <- "ctrl"
metadata$sample[which(str_detect(metadata$cells, "^stim_"))] <- "stim"最终的metadata,包含了用来质控的指标:

# 将新的metadata重新添加回seurat
[email protected] <- metadata
#保存
save(merged_seurat, file="data/merged_filtered_seurat.RData")评估质量指标
我们将对多个质量指标进行控制并去除不合格的细胞,这些指标包括:
细胞计数(
Cell counts)每个细胞的UMI(
UMI counts per cell)每个细胞观察到的基因(
Genes detected per cell)UMIs vs. genes detected线粒体counts比例(
Mitochondrial counts ratio)Novelty
什么是doublets?简单的说就是两个细胞混在一起,可能发生在细胞捕获过程中,并且可能会误导认为是两种细胞类型的过渡态(transitory states),所以应该去除(单细胞预测Doublets软件包汇总-过渡态细胞是真的吗?)。
我们为什么不检查doublets呢?许多的工作流程都是通过设置UMI或genes的最大阈值进行的,其原理为大量的reads或基因表明存在着多个细胞,尽管此理由似乎很直观,但并不准确。Scrublet(https://github.com/AllonKleinLab/scrublet)是检测doublets的重要工具,但是我们还没有对它进行基准测试。我们不建议使用阈值进行筛选,当我们确定了每个cluster的marker后,我们建议探索这些标记,以确定这些marker是否适用于一种以上的细胞类型。
细胞计数
细胞计数由检测到的barcode的数量确定。对于此实验,预计将有12,000-13,000个细胞。inDrops的细胞捕获效率略高(70-80%),而10X则为50-60%。注意:如果用于文库制备的细胞浓度不准确,捕获效率可能会低得多。我们不应通FACS或Bioanalyzer确定细胞浓度(这些工具无法准确确定浓度),而应使用血细胞计数器或自动细胞计数器来计算细胞浓度。
同时在inDrops和10X中,也会发生得到的细胞数目(即细胞barcodes数)大于细胞数的情况。
# Visualize the number of cell counts per sample
metadata %>%
ggplot(aes(x=sample, fill=sample)) +
geom_bar() +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) +
theme(plot.title = element_text(hjust=0.5, face="bold")) +
ggtitle("NCells")
从上图可以看出每个sample的细胞数均在15000以上。
每个细胞的UMI
每个细胞的UMI计数通常应高于500,500是预期的下限。如果UMI在500-1000计数之间,虽然可以使用,但可能应该对细胞进行更深的测序。
# Visualize the number UMIs/transcripts per cell
metadata %>%
ggplot(aes(color=sample, x=nUMI, fill= sample)) +
geom_density(alpha = 0.2) +
scale_x_log10() +
theme_classic() +
ylab("Cell density") +
geom_vline(xintercept = 500)
我们可以看到大多数细胞的UMI数均在1000以上。
每个细胞观察到的基因
正常情况应该和UMI相同,也是有一个大大的峰,如果我们在主峰的右边也看到了一个小峰,出现的原因也是多种多样的,可能是由于捕获单个细胞失败,或者具有某些生物学意义,需要仔细评估。
对于高质量数据,比例直方图应包含一个大峰,该峰代表封装的细胞。如果我们在主要峰的右侧看到一个小峰(我们使用的数据中不存在这种情况),或者细胞的双峰分布,则可能表明有两件事。一个可能是某些原因导致一组细胞失败。一个可能是存在生物学上不同类型的细胞(例如,静止细胞群体,不太复杂的目标细胞),和/或一种类型比另一种类型小得多(即,具有高计数的细胞可能是尺寸较大的细胞) )的情况。
因此应该使用本课中介绍的其他指标来评估此阈值。
# Visualize the distribution of genes detected per cell via histogram
metadata %>%
ggplot(aes(color=sample, x=nGene, fill= sample)) +
geom_density(alpha = 0.2) +
theme_classic() +
scale_x_log10() +
geom_vline(xintercept = 300)
# Visualize the distribution of genes detected per cell via boxplot
metadata %>%
ggplot(aes(x=sample, y=log10(nGene), fill=sample)) +
geom_boxplot() +
theme_classic() +
theme(axis.text.x = element_text(angle = 45, vjust = 1, hjust=1)) +
theme(plot.title = element_text(hjust=0.5, face="bold")) +
ggtitle("NCells vs NGenes")

UMIs vs. genes
通常我们会一起评估UMI的数量和每个细胞检测到的基因数量。下图是通过线粒体reads的分数绘制的基因数和UMI数之间的关系,显示出线粒体reads分数在带有低基因数的低计数细胞中很高。
为什么我们需要去除线粒体基因占比较高的细胞呢,主要是由于破损细胞的胞质中的mRNA会由于细胞破损游离出来,而线粒体中的被保留了下来,线粒体基因比例高。这样的细胞可以通过count数和基因数联合去除。
质量较差的细胞的基因和UMI可能很低,对应于该图左下象限中的数据点。好的细胞通常会同时显示每个细胞更多的基因和更高数量的UMI。
通过此图,我们还可以评估线的斜率以及该图右下象限中数据点的分布。具有大量的UMI,但只有少数基因的细胞可能是垂死的细胞,也可能是低复杂度的细胞类型(即红细胞)。
# Visualize the correlation between genes detected and number of UMIs and determine whether strong presence of cells with low numbers of genes/UMIs
metadata %>%
ggplot(aes(x=nUMI, y=nGene, color=mitoRatio)) +
geom_point() +
scale_colour_gradient(low = "gray90", high = "black") +
stat_smooth(method=lm) +
scale_x_log10() +
scale_y_log10() +
theme_classic() +
geom_vline(xintercept = 500) +
geom_hline(yintercept = 250) +
facet_wrap(~sample)
线粒体counts比例
我们可以识别死细胞或垂死细胞是否存在大量线粒体污染。除非必要,我们将线粒体比率超过0.2的细胞定义为线粒体计数质量差的样品。
# Visualize the distribution of mitochondrial gene expression detected per cell
metadata %>%
ggplot(aes(color=sample, x=mitoRatio, fill=sample)) +
geom_density(alpha = 0.2) +
scale_x_log10() +
theme_classic() +
geom_vline(xintercept = 0.2)
复杂度
其实进行少量测序的每个细胞具有高度的复杂性,这主要是由于我们没有对每个样品进行基因深度测序。样品中的异常细胞可能其RNA复杂度较低,我们可以通过该指标检测筛选低复杂度(如红细胞)的污染。一般情况下,我们把评分(novelty score)设置为0.8。
# Visualize the overall complexity of the gene expression by visualizing the genes detected per UMI
metadata %>%
ggplot(aes(x=log10GenesPerUMI, color = sample, fill=sample)) +
geom_density(alpha = 0.2) +
theme_classic() +
geom_vline(xintercept = 0.8)
NOTE: Reads per cell is another metric that can be useful to explore; however, the workflow used would need to save this information to assess. Generally, with this metric you hope to see all of the samples with peaks in relatively the same location between 10,000 and 100,000 reads per cell.
过滤
线粒体数相对高的细胞也可能参与了呼吸过程,是某研究中正需要的,因此作者认为在设置条件时应综合考虑,尽量宽松,防止有意义的生物学变化被删除。
细胞层面过滤
nUMI > 500
nGene > 250
log10GenesPerUMI > 0.8
mitoRatio < 0.2
用subset()函数对Seurat对象进行设置:
# Filter out low quality reads using selected thresholds - these will change with experiment
filtered_seurat <- subset(x = merged_seurat,
subset= (nUMI >= 500) &
(nGene >= 250) &
(log10GenesPerUMI > 0.80) &
(mitoRatio < 0.20))基因层面过滤
删除0表达值的基因和在少于10个细胞中表达的基因。
# Output a logical vector for every gene on whether the more than zero counts per cell
# Extract counts
counts <- GetAssayData(object = filtered_seurat, slot = "counts")
# Output a logical vector for every gene on whether the more than zero counts per cell
nonzero <- counts > 0
# Sums all TRUE values and returns TRUE if more than 10 TRUE values per gene
keep_genes <- Matrix::rowSums(nonzero) >= 10
# Only keeping those genes expressed in more than 10 cells
filtered_counts <- counts[keep_genes, ]
# Reassign to filtered Seurat object
filtered_seurat <- CreateSeuratObject(filtered_counts, meta.data = [email protected])Re-assess QC metrics
过滤之后可以回顾一下过滤前后的效果,以评判过滤指标是否合适。
Saving filtered cells
# Create .RData object to load at any time
save(filtered_seurat, file="data/seurat_filtered.RData")(五)Count Normalization and Principal Component Analysis
获得高质量的单细胞后,单细胞RNA-seq(scRNA-seq)分析工作流程的下一步就是执行聚类。聚类的目标是将不同的细胞类型分成独特的细胞亚群。为了进行聚类,我们确定了在细胞之间表达差异最大的基因。然后,我们使用这些基因来确定哪些相关基因集是造成细胞之间表达差异最大的原因。
数值归一化
归一化最重要的目的就是使表达水平在细胞之间和/或细胞内更具有可比性。那么在归一化中主要需要处理的因素包括:
测序深度:考虑测序深度是比较细胞之间基因表达的必要条件。在下面的示例中,每个基因在细胞2中的表达似乎都增加了一倍,但这是细胞2具有两倍测序深度的结果。
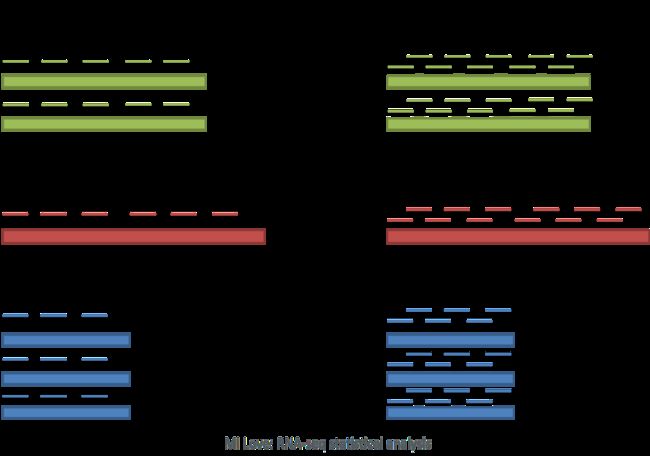
因此,要准确比较细胞之间的表达,有必要对测序深度进行归一化。
基因长度:需要基因长度来比较同一细胞内不同基因之间的表达。比对到较长基因的reads数似乎与较高表达的较短基因具有相同的计数/表达。

如果进行的5’末端或3’末端的测序,则不需要考虑基因长度的影响;
如果使用全长测序则需要考虑。
主成分分析(PCA)
PCA是一个用来强调差异和相似性的技术(一文看懂PCA主成分分析),这里推荐一个学习视频:StatQuest's video(https://www.youtube.com/watch?v=_UVHneBUBW0)
下面是PCA的示例模拟过程,帮助理解:
如果你已经定量了两个样本(或细胞)中四个基因的表达,则可以绘制这些基因的表达值,其中一个样本在x轴上表示,另一个样本在y轴上表示,如下所示:

我们可以沿代表最大变化的方向在数据上画一条线,在此示例中为对角线。数据集中的最大变异是在组成两个端点的基因。我们还看到基因在该线的上方和下方有些不同。我们可以在数据上绘制另一条线,代表数据中变化第二大的变量。

末端附近的基因将是变异最大的基因。这些基因在数学上对线的方向影响最大。

例如,基因C值的微小变化将极大地改变较长线的方向,而基因A或基因D的微小变化对其几乎没有影响。

我们还可以旋转整个图,保证线条方向是从左到右和从上到下。现在,可以将这些线视为代表变化的轴。这些轴本质上是“主要组分”,其中PC1代表数据的最大差异,PC2代表数据的第二大差异。

如果有N个细胞,以此类推。。。
确定PCs后,则需要对每个PC进行评分,按照以下步骤对所有样本PC对(sample-PC pairs)计算分数:
(1)首先,根据基因对每个PC的影响程度,为其分配“影响力”评分。对给定PC没有任何影响的基因得分接近零,而具有更大影响力的基因得分更高。PC线末端的基因将产生更大的影响,因此它们将获得更大的分数,但两端的符号相反。

(2)确定影响分数后,使用以下公式计算每个样本的分数:
Sample1 PC1 score = (read count * influence) + ... for all genes以我们的2个样本示例,以下是分数的计算方式:
## Sample1
PC1 score = (4 * -2) + (1 * -10) + (8 * 8) + (5 * 1) = 51
PC2 score = (4 * 0.5) + (1 * 1) + (8 * -5) + (5 * 6) = -7
## Sample2
PC1 score = (5 * -2) + (4 * -10) + (8 * 8) + (7 * 1) = 21
PC2 score = (5 * 0.5) + (4 * 1) + (8 * -5) + (7 * 6) = 8.5(3)一旦为所有PC计算了这些分数,就可以将其绘制在简单的散点图上。下面是示例图:

对于具有大量样本或细胞的数据集,通常会绘制每个样本/细胞的PC1和PC2分数。由于这些PC解释了数据集中最大的变化,因此更相似的样本/细胞将在PC1和PC2聚在一起。请参见下面的示例:

Image credit: https://github.com/AshwiniRS/Medium_Notebooks/blob/master/PCA/PCA_Iris_DataSet.ipynb
对于我们的单细胞数据,我们最终会选择10-100 PC去对细胞进行聚类分析,而不是全部基因。
未完待续。
你可能还想看
如何使用Bioconductor进行单细胞分析?
对一篇单细胞RNA综述的评述:细胞和基因质控参数的选择
如何火眼金睛鉴定那些单细胞转录组中的混杂因素
什么?你做的差异基因方法不合适?
Seurat亮点之细胞周期评分和回归
送书
在上周的留言送书活动中,恭喜下面这位读者获得书籍“Python:自动化测试实战”,请及时与生信宝典编辑(shengxinbaodian)联系。

本期的留言主题是:您近期最喜欢的文章是哪一篇呢?是关于什么内容的呢?欢迎转发朋友圈并留言评论。
因有读者反馈刷赞一事,因此更改送书规则为在留言中选择“有缘人”获得下面由北京大学出版社赞助的书籍(联系小编时请附上分享截图),内容契合留言主题且实际留言得赞越高者越可能成为“有缘人”,欢迎分享并互相监督,结果在下一期送书活动中公布:

本书参与了京东活动每满100减50,活动时间是6.16-6.18,心动可先行动~
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


