探索seurat包
- 一、了解包的基本内容
- 1. 加载
- 2. 基本信息
- 二、使用
- 1. 导入数据
- 2. 创建对象和基本操作
- 1)检查数据
- 2)创建对象
- 3)查看对象内容
- 4)调用对象内容
- 5)向对象插入内容
- 6)可视化对象内容
- 3. 归一化
- 4. 挑选高变异基因
- 5. 标准化及去除混杂因素影响
- 6. 降维
- 1)PCA
- 2)UMAP
- 3)tSNE
- 7. 聚类
- 8. 确定差异表达基因
- 9. 总结
一、了解包的基本内容
1. 加载
if(!require(Seurat))
BiocManager::install('Seurat')
if(!require(scRNAseq))
BiocManager::install('scRNAseq')
2. 基本信息
-
参考
https://satijalab.org/seurat/v3.1/pbmc3k_tutorial.html -
输入数据
a. cellranger 输出的10X数据
b. 未经过标准化的数据:raw counts /TPMs
二、使用
1. 导入数据
data(fluidigm)
assay(fluidigm) <- assays(fluidigm)$rsem_counts
ct <- floor(assays(fluidigm)$rsem_counts)
2. 创建对象和基本操作
1)检查数据
meta <- as.data.frame(colData(fluidigm)) #细胞相关信息
counts <- ct #表达矩阵
identical(rownames(meta),colnames(counts))#检查counts的列名和meta的行名是否保持一致
fivenum(apply(ct,1,function(x) sum(x>0))) #基因表达的细胞数目
fivenum(apply(ct,2,function(x) sum(x>0) ))#细胞表达的基因数目

由上图可见有至少25%的基因不表达,至少50%的基因表达的细胞数目不超过5个;细胞表达的基因数目多于1418个
2)创建对象
sce <- CreateSeuratObject(counts = counts, #表达矩阵
meta.data =meta, #细胞信息
min.cells = 3, #基因表达的最少细胞数目
min.features = 200,#细胞表达的最少基因数目
project = "sce") #项目名称

过滤了11599个基因和0个细胞
3)查看对象内容
str(sce)



在后续操作时默认使用active.assay,也可以使用assay参数指定使用的表达矩阵
4)调用对象内容
head(sce@assays$RNA@counts[,1:4])

sce存储的表达矩阵是类矩阵,只能使用Matrix包的函数进行操作
5)向对象插入内容
为元信息增加线粒体基因的比例,如果线粒体基因所占比例过高,意味着这可能是死细胞
mito.genes <- grep(pattern = "^MT-", x = rownames(x = sce@assays$RNA@data), value = TRUE) #这个例子的表达矩阵里面没有线粒体基因
percent.mito <- Matrix::colSums(sce@assays$RNA@counts[mito.genes, ]) / Matrix::colSums(sce@assays$RNA@counts)
sce <- AddMetaData(object = sce, metadata = percent.mito,col.name = "percent.mito")


在 meta.data元素里新增了一列
6)可视化对象内容
a. 小提琴图
VlnPlot(object = sce, features = c("nCount_RNA", "nFeature_RNA", "percent.mito"), group.by = 'Biological_Condition')

不同类型的细胞测序深度和表达的基因数目均有差异
b. 散点图
tail(sort(Matrix::rowSums(sce@assays$RNA)))#高表达基因

plot1 <- FeatureScatter(sce, feature1 = "SOX11", feature2 = "EEF1A1",group.by = "Biological_Condition") #基因间关系
plot2 <- FeatureScatter(sce, feature1 = "nCount_RNA", feature2 = "nFeature_RNA",group.by = "Biological_Condition") #细胞信息间关系
CombinePlots(plots = list(plot1, plot2)) #展示在同一页面

3. 归一化
sce1<-sce #备份
sce <- NormalizeData(object = sce,
normalization.method = "LogNormalize", #文库大小归一化及log
scale.factor = 10000 #文库大小
)

归一化后的表达矩阵存储在data元素里
4. 挑选高变异基因
使用数据:counts,而不是data
sce <- FindVariableFeatures(sce, selection.method = "vst", nfeatures = 2000)

2000个高变异基因存储在var.features元素里
优化函数:sctransform
- 可视化
plot1 <- VariableFeaturePlot(sce)
top10 <- head(VariableFeatures(sce), 10) #挑选变化最大的10个基因
plot2 <- LabelPoints(plot = plot1, points = top10, repel = TRUE,xnudge=0,ynudge=0) #标注基因名
CombinePlots(plots = list(plot1, plot2),legend = "bottom")

5. 标准化及去除混杂因素影响
a. seurat包要求PCA之前必须标准化
b. 使用数据:data
sce <- ScaleData(sce,
features = NULL,#默认使用高变异基因进行标准化
#vars.to.regress="nUMI"#去除文库大小的影响
)
sce[["RNA"]]@scale.data[1:4,1:4]

标准化后的矩阵存储在scale.data元素里
6. 降维
a. 使用高变异基因进行降维,从而可以加快计算速度
b. 降维结果存储在reductions元素里
c. 使用少量的PC既能关注到关键的生物学差异,又能够不引入更多的技术差异,相当于一种保守性的做法
d. 使用scale.data元素
1)PCA
sce <- RunPCA(sce,
features = VariableFeatures(object = sce),#使用高变异基因降维
ndims.print=1:2,nfeatures.print=5 #打印出来的维度和基因数目
)


- 可视化
a. 基因对每个主成分的重要性占比情况
VizDimLoadings(sce, dims = 1:2, reduction = "pca")

b. 主成分图
DimPlot(sce, reduction = "pca",group.by = "Biological_Condition")
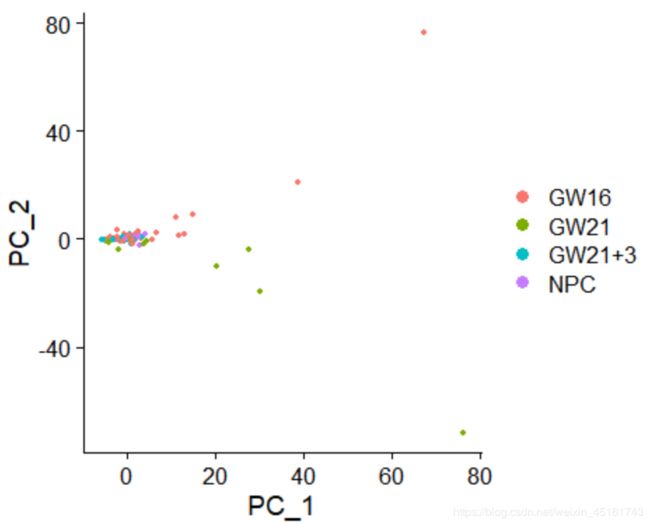
NPC和GW细胞完全混合在一起,说明挑选高变异基因的方法不对,导致数据分布与生物学认知不同,按道理来说应该要更换方法重新挑选高变异基因,但因为只是探索所以用该套数据继续往下做
c. 热图
DimHeatmap(sce, dims = 1, balanced = TRUE)
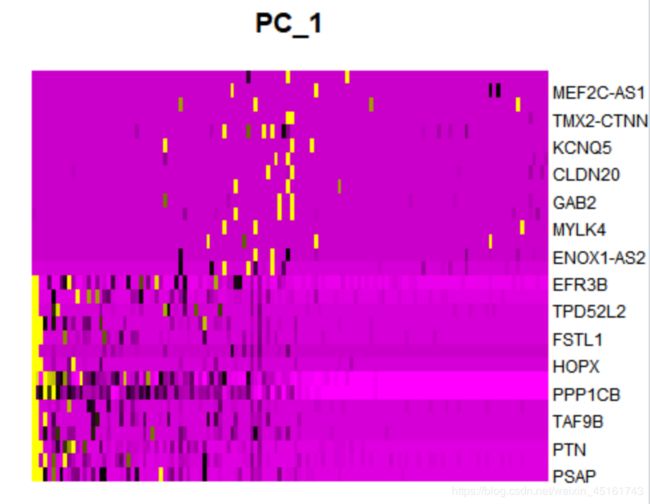
主成分越前的贡献基因按道理来说在样本里的差异性越大,但是因为挑选的高变异基因不太对,所以体现不出这种差异
正确图:

d. 挑选主成分
ElbowPlot(sce)

一般取点保持平衡趋势的前几个主成分,在上图应取前20个主成分
2)UMAP
sce <- RunUMAP(sce, dims = 1:20) #取PCA前20个主成分进行计算,加快速度
DimPlot(sce, reduction = "umap",group.by = "Biological_Condition")


说明该套数据存在技术变异,影响了可视化结果。从文献中可知该套数据使用了两种测序深度,所以可以推测进行下游分析前需去除测序深度变异(目前没有找到一个细胞属性可以很好地进行批次处理)
3)tSNE
sce <- RunTSNE(object = sce, dims.use = 1:20)
DimPlot(sce, reduction = "tsne",group.by = "Biological_Condition")


效果没有umap好
7. 聚类
- 确定邻域
sce <- FindNeighbors(sce, dims = 1:20)
存储在graph元素里

- 分类
sce <- FindClusters(sce,resolution = 1.2) #resolution值越大分组越多
head(Idents(sce), 5)


存储在meta.data元素里
8. 确定差异表达基因
运行速度很慢
- 仅确定某类
marker1<-FindMarkers(sce, ident.1 = 1)
head(marker1)

- 确定所有类
marker<-FindAllMarkers(sce)

- 可视化
- 在降维图上可视化基因表达
FeaturePlot(sce, features ="RPL18AP3" )

- 热图
library(dplyr)
top10 <- marker %>% group_by(cluster) %>% top_n(n = 10, wt = avg_logFC)
DoHeatmap(sce, features = top10$gene) + NoLegend() #无图例

- 在降维图上进行注释
new.cluster.ids <-top10$gene
names(new.cluster.ids)<-rep(0:3, each = 10)
sce <- RenameIdents(sce, new.cluster.ids)


只使用了new.cluster.ids中每一类的第一个基因修改ident
DimPlot(sce, reduction = "pca", label = TRUE, pt.size = 0.5) + NoLegend()

9. 总结
a. 创建对象所需元素: counts矩阵
b. 归一化函数
NormalizeData :文库大小归一化和log化
ScaleData :标准化和技术差异校正
c. 挑选基因函数
FindVariableGenes :挑选适合进行下游分析的基因集
FindAllMarkers:挑选各个亚群的标志基因
d. 降维函数: RunPCA,RunTSNE,RunUMAP
e. 可视化基因函数:
FeaturePlot:不同基因在所有细胞的表达量
VlnPlot:不同基因在不同分群的表达量
DoHeatmap:选定基因集绘制热图