多组学分析,以其数据维度和工作量之大,一直经久不衰,在经过2019年的免疫热后,2020年生信要怎么玩呢?今天给大家提供一个思路,对 就是多组学。话不多说,来看正文。

The Integrated Genomic Landscape of Thymic Epithelial Tumors.
期刊:Cancer cell
日期:2018-02-12
DOI :10.1016/j.ccell.2018.01.003
文章背景:
胸腺上皮肿瘤( TETs )是最罕见的成人恶性肿瘤之一。在TET中,胸腺瘤是最主要的,其特点是与自身免疫性疾病有独特的联系,其次是胸腺癌,后者较不常见,但在临床上更具侵略性。通过对117种TET进行多平台组学分析,我们定义了这些肿瘤的四个亚型,这些亚型由基因组标志以及与生存和世界卫生组织组织学亚型的关联定义。我们进一步证明了胸腺瘤特异性突变致癌基因GTF2I的显着患病率,并探讨了其对多平台分析的生物学影响。我们进一步观察到 HRAS 中突变的富集,NRAS和TP53。最后,我们确定了胸腺瘤与自身免疫性疾病重症肌无力之间的分子联系,其特征是肿瘤中的肌肉自身抗原过表达和非整倍性增加。
意义:
总而言之,这项工作代表了迄今为止对胸腺上皮肿瘤的最大,最全面的分子分型分析。
数据来源:
117对肿瘤血液样本的外显子测序数据(来自TCGA THYM研究,我们是拿不到的)
收集并分析TCGA其他21种肿瘤的CNV数据(不要被数据量吓到,其实作者就是用TCGA数据分析了一下TMB)
mRNA-seq数据(测了几个看完文章也没看出来)
117例miRNA-seq数据(文章只用了其中的一部分)
117例DNA甲基化数据(Illumina Infinium HM450 阵列)
反向蛋白阵列数据(从200例样品中最后筛剩下90例)
临床资料(来自世界卫生组织)
分析流程:
结果展示:
在对117组 tumor-normal配对样本的全外显子测序数据进行筛选后,剩余100对样本数据被用来识别显著的体细胞突变(图1)。他们用MutSig2CV方法识别到了四个显著突变的基因: GTF2I, HRAS, TP53, and NRAS ,其中GTF2I是突变频率最高、最显著的基因,也是已知的TET突变基因;PyClone的克隆分析表明这四个基因更易在发病期或肿瘤发展前期发生突变。在与TCGA中的另外21中癌症谱进行对比时,发现TET具有比较低的突变频率 (图2),而且大多数患者没有发生体细胞拷贝数变异(sCNA);对 RNA-seq数据的分析也没有发现与TETs相关的反复性融合事件以及病毒/细菌成分。
作者整合了多个TCGA平台的聚类结果,通过改进后的COCA (2012年发表的一篇文章)方法识别出了TETs不同的分子亚型。综合sCNV, mRNA, miRNA, DNA甲基化和反向蛋白阵列(RPPA)数据得到了一个模糊赋值的加权矩阵用来识别分子亚型,最终基于WHO的分型得到了4种TETs亚型:第一种主要由B型代表,第二种2主要由TC型代表,第三种主要是AB型,第四种是A型和AB型的混合(图3B),第一种亚型中的病例也多患有MG;生存分析表明在这四种亚型中,第二种的整体生存状况较差,这与事实相符;此外,作者还发现第1、3重亚型患者淋巴细胞含量较高,GTF2I突变主要发生在3、4种亚型中, HRAS 突变主要发生在第四种亚型中GTF2I, a thymoma specific oncogene:
由于GTF2I突变在A型和AB型胸腺瘤中具有高度一致性,因此作者利用多平台数据来进一步区分A型和AB型。他们发现所有的突变都发生在L424H,这可能与GTF2I基因突变是一致的;GTF2I突变体在细胞形成、受体激酶信号转导、酸性受体、神经元生成以及WNT和SNH信号通路中均有较高的基因表达;对RPPA 数据的多重假设检验发现91个蛋白在GTF2I突变体中显著下调;通路分析表明在GTF2I突变体中细胞凋亡、细胞周期、DNA损伤反应、激素受体信号、乳腺激素信号转导、激素受体信号转导途径等的表达较低。


图3。来自五个数据平台的子类型的集成无监督聚类(A)共识聚类将TET样品分为四个分子亚型(n = 117)。顶部的蓝色和白色热图显示了示例共识。中心的蓝色和黄色热图显示了与每个单独的数据类型集群成员质心的相关性。底部显示了四个显着突变的基因之一中突变的存在(黑色)或不存在(灰色)。(B)按WHO组织学亚型分类的各组样本摘要。(C)跨分子亚型的生存差异。第3组中的样本缺少生存数据。(D)从TumorMap产生的样品的图谱按病理状态着色。根据相似性将样品放置在整合所有平台的基因组图谱中。对于每个群集,单平台标记在细线上方列出,而PARADIGM结果在细线下方列出。另请参见图S3-S5。
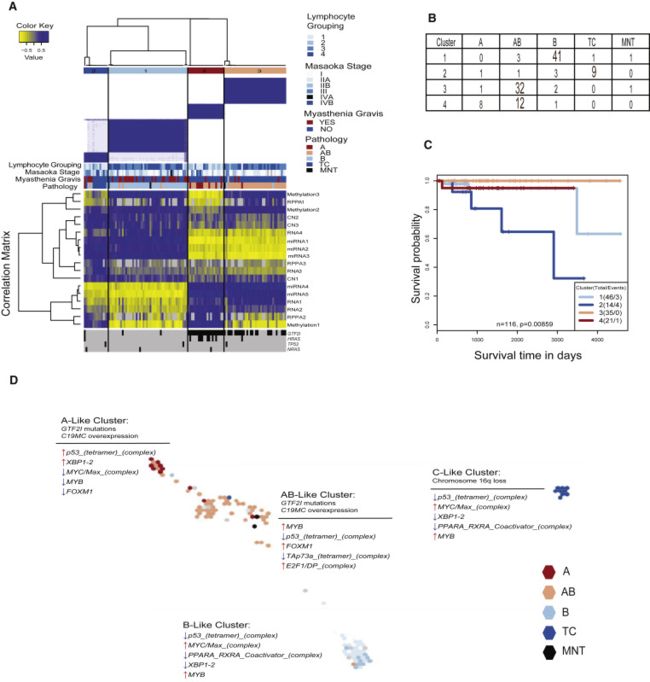
图4。胸腺瘤特异性癌基因GTF2I的多平台分析
(A)GTF2I的棒棒糖图,显示了在GTF2I中观察到的所有突变。绿框标记了类似GTF2I的重复区域。
(B)与TET相比,其他癌症谱系中GTF2I 的体细胞突变频率。
(C)在A型和AB型胸腺瘤中的 GTF2I 突变基因表达特征。
(D)A和AB型的 GTF2I 甲基化标记。根据RNA序列数据,还报告了10个分类错误的样品。
(E)GTF2I 突变(红色)和野生型(蓝色)肿瘤的途径得分的箱线图。箱线图显示中位数,上四分位数和下四分位数,胡须表示四分位数范围,离群值用点标记,p值基于ANOVA测试。

图5。sCNA的模式和与自身免疫相关的基因表达(A和B)比较了跨组织学类型(A)的所有样本以及仅B1,B2,B3样本的子集的重症肌无力的阳性(MG +)和阴性(MG-)状态的染色体臂发生率组织学类型(B)。对于箱线图:框中的线表示中位数;下部和上部铰链分别与第一和第三四分位数相对应;上下晶须延伸至1.5倍四分位间距;离群数据显示为点。(C)具有B1,B2,B3组织学的样本的基因水平sCNA频率分布图,比较重症肌无力状态的历史。的χ 2独立的测试施加到臂级SCNA每个染色体臂,以确定MG + MG-和状态之间显著富集事件。小于0.05(-log q值= 3)的错误发现率(q)显示为增益(红色)和缺失(蓝色)。(D)选定差异表达基因的Log2归一化基因表达。
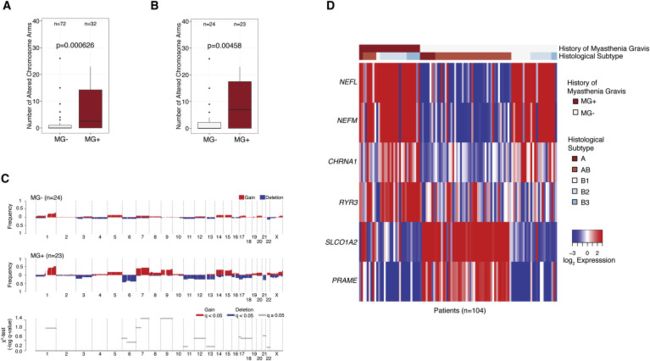
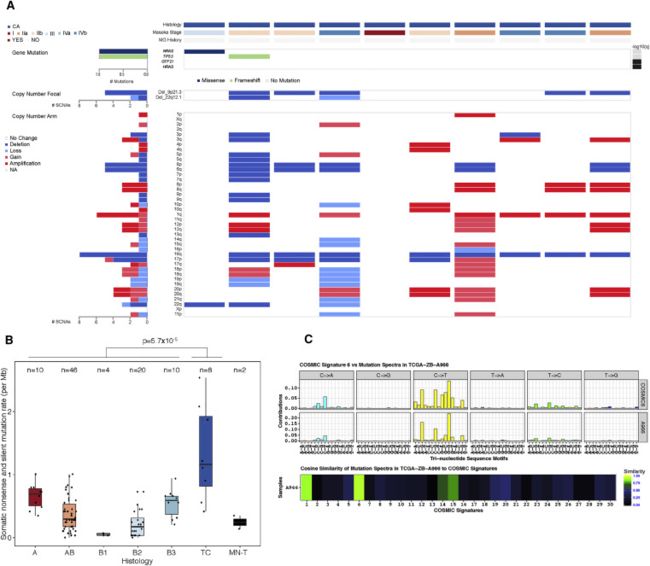
图6。胸腺癌的基因组分析(A)TC型肿瘤中DNA突变的情况。面板顶部的矩阵描述了临床信息。面板的中心描绘了TC类型的肿瘤中的个别突变,这些突变按突变类型进行了颜色编码,用于先前确定的显着突变的基因和病灶拷贝数变化。面板的下半部分描述了按CNA类型编码的每个样本颜色的手臂级sCNA。左侧的条形图描述了每个样本的总sCNA数。(B)通过组织学展示样品TMB的箱线图。方框中的线表示中位数;下部和上部铰链分别与第一和第三四分位数相对应;上下晶须延伸至1.5倍四分位间距;离群数据显示为点。为了避免歪曲结果,排除了1个超突变TC样本和1个TMB = 0的TC样本。(C)COSMIC签名6(微卫星不稳定肿瘤)的归一化谱图和超突变TC样品(TCGA-ZB-A966)的SNV突变谱,沿96个碱基的替换类型在三核苷酸 序列基序中(顶部)和TCGA-ZB-A966中的突变谱与30个策划的COSMIC签名之间的余弦相似度。
文章亮点:
• 通过多组学数据(sCNV, mRNA, miRNA, DNA甲基化和反向蛋白阵列(RPPA)数据)构建出四种与生存相关(预后)的稳健的分子TET亚型。
• 在成人癌症中,胸腺瘤的突变负担最低。(突变负担,就是TMB(tumor mutation burden),不要换了个词就不认识了..)
• 观察到HRAS,NRAS,TP53和复发性GTF2I突变的富集。
• 自身免疫靶标的表达和非整倍性将胸腺瘤与重症肌无力联系起来。
其实看懂别人的文章并不难,难的是自己怎么用起来,看完全文之后我不认为这是一个好模仿的套路,作者用了很多自己的测序数据和大量我们拿不到的数据。
那么怎么用TCGA数据去操作多组学分析呢?不妨参考之前的一篇帖子 多组学生信分析汇总
