PlantCom:基于内参的宿主相关丰度定量分析揭示植物根系微生物组的微生物总量变化
文章目录
- 基于内参的宿主相关丰度定量分析揭示植物根系微生物组的微生物总量变化
- 摘要
- 背景介绍
- 图1.植物根部微生物组定量丰度分析的优势和实验步骤
- 结果
- spike-in内参质粒和HA-QAP方法的原理
- 内参质粒(spike-in)适用于定量分析微生物组谱
- 图2. 在细菌模拟实验中HA-QAP比传统的基于相对丰度的分析更准确
- 在人工混菌实验中检测微生物总量以评估方法的设计和合理性
- HA-QAP在模拟实验中检测根微生物组的微生物总量
- HA-QAP可在模拟实验中准确检测单个根微生物组成员的变化
- 图3. 在真菌模拟实验中HA-QAP比基于相对丰度的经典分析更为准确
- HA-QAP方法可检测自然根样品中微生物总量的变化
- 图4. HA-QAP揭示了自然根样品中微生物总量的降低
- 微生物总量是水稻根系微生物组对干旱胁迫响应变化的关键特征
- 图5. HA-QAP显示干旱胁迫下水稻根系的微生物总量增加
- HA-QAP显示感染根腐病的小麦根际微生物总量绝对增加
- 图6. HA-QAP显示出患有根腐病的小麦植株中微生物总量增加
- 讨论
- Reference
- 猜你喜欢
- 写在后面

基于内参的宿主相关丰度定量分析揭示植物根系微生物组的微生物总量变化
Host-Associated Quantitative Abundance Profiling Reveals the Microbial Load Variation of Root Microbiome
Plant Communications
Impact Factor: NA
https://doi.org/10.1016/j.xplc.2019.100003
发表日期:Volume 1, Issue 1, 13 January 2020, 100003
第一作者:Xiaoxuan Guo(郭晓璇)1,2, Xiaoning Zhang(张小宁)1,2,3, Yuan Qin(秦媛)1,2,3
通讯作者:Xiangdong Fu(傅向东)4,5 ([email protected]) 和 Yang Bai(白洋)1,2,3([email protected])
合作作者:Yong-Xin Liu(刘永鑫)1,2, Jingying Zhang(张婧赢)1,2,
Na Zhang(张娜)1,2,3, Kun Wu4,5, Baoyuan Qu(曲宝原)1,2, Zishan He(贺子姗)1,2,3, Xin Wang(王鑫)1,2, Xinjian Zhang6,
Ste´ phane Hacquard7
主要单位:
- 中国科学院,种子创新研究院,遗传与发育生物学研究所,植物基因组学国家重点实验室 (State Key Laboratory of Plant Genomics, Institute of Genetics and Developmental Biology, Innovation Academy for Seed Design, Chinese Academy of Sciences
(CAS), Beijing 100101, China) - 中国科学院遗传与发育生物学研究所,中国科学院-英国约翰英纳斯中心植物和微生物科学联合研究中心(CAS-JIC Centre of Excellence for Plant and Microbial Science (CEPAMS), Institute of Genetics and Developmental Biology, Chinese Academy of Sciences (CAS), Beijing 100101, China)
- 中国科学院大学,现代农学院 (University of Chinese Academy of Sciences, College of Advanced Agricultural Sciences, Beijing 100049, China)
- 中国科学院,种子创新研究院,遗传与发育生物学研究所,植物细胞与染色体工程国家重点实验室 (State Key Laboratory of Plant Cell and Chromosome Engineering, Institute of Genetics and Developmental Biology, Innovation Academy for Seed Design, Chinese Academy of Sciences (CAS), Beijing 100101, China)
- 中国科学院大学,生命学院 (College of Life Sciences, University of Chinese Academy of Sciences, Beijing, 100049, China)
- 齐鲁工业大学生态研究所,山东省应用微生物学重点实验室 (Shandong Provincial Key Laboratory of Applied Microbiology, Ecology Institute, Qilu University of Technology (Shandong Academy of Sciences), Jinan 250014, China)
- 马克斯·普朗克植物育种研究所 (Max Planck Institute for Plant Breeding Research, Cologne 50829, Germany)
新闻稿
- Plant Com:中科院遗传发育所白洋组开发定量检测宿主微生物组的HA-QAP技术(王二涛点评)
摘要
与植物相关的微生物对于植物在自然环境条件下的生长和生存至关重要。迄今为止,涉及高通量扩增子测序的微生物组研究几乎都集中在微生物分类群的相对丰度上。但是,该技术不能评估微生物总量(microbial load)和单个微生物相对于宿主植物组织数量的丰度。如果样品之间的微生物总量发生很大变化,则基于微生物相对丰度的群落结构定量方法将会对微生物组与植物生理状态之间相互作用的分析有很大限制。在这里,我们建立了基于内参的定量丰度分析(HA-QAP)工作流程,以准确检测微生物总量和单个根系微生物组成员相对于宿主植物根源DNA的定殖。我们分别使用模拟实验,扰动实验以及宏基因组测序验证了HA-QAP方法的可行性和准确性。与基于微生物相对丰度的经典方法相比,HA-QAP方法降低了假阳性和假阴性,并揭示了根微生物组相对宿主植物的总量。用HA-QAP方法,我们发现在健康水稻和小麦的根微生物组样品中,细菌标记16S rRNA基因与植物基因组的拷贝数比范围为1.07至6.61,真菌ITS的微生物标记基因范围为0.40至2.26。此外,使用HA-QAP,我们发现微生物总量的增加代表暴露于干旱胁迫的水稻植物和患有根腐病的小麦植物的根微生物组变化的关键特征,这显著影响差异分类单元和物种互作网络的模式。鉴于其准确性和技术可行性,HA-QAP将有助于我们了解根微生物组与植物之间的真正相互作用。
微生物总量(microbial load):定义在一个对象或物种中包括污染微生物的数量和类型。 (Definition The number and type of microorganisms contaminating an object or organism)
背景介绍
健康植物的根部被复杂多样的微生物群落(微生物组)所定殖,这些微生物群落在植物养分获取、胁迫耐受性和抗病性中发挥着关键作用。与未种植的土壤相比,根际微生物会增加10–100倍。细菌16S rRNA基因或真菌内部转录间隔区(ITS)的高通量扩增子测序已在几种模式植物和农作物中用于检测根部微生物组的组成和基于微生物相对丰度的变异,已有的研究揭示了根部微生物组的组成主要是受地理位置、土壤类型、非生物和生物胁迫以及宿主基因型的影响。随着根微生物组-宿主植物相互作用的被关注日益增加,植物健康与植物微生物总量之间的相互作用亟待研究。
当前的微生物组分析方法是基于细菌或真菌的相对丰度(称为经典分析方法;请参见下文),它们无法揭示相对于宿主植物组织丰度的实际微生物总量(图1a)。在微生物总量未知的前提下,我们无法确定某些物种的富集是由于其绝对丰度的增加还是其他优势物种相对丰度的降低所致。相应地,由于它是基于相对丰度去下结论(图1b,c),因此经典的剖析方法会产生假阴性和假阳性结果。例如,植物根部B中的微生物与根部A中的微生物具有相同的比例组成,但总微生物量却是后者两倍。经典的分析方法在根A和根B中检测到相同的微生物组(具有相同的微生物组成),从而导致假阴性结果(图1b,c)。在另一个典型示例中,根C与根A相比,单个微生物的水平没有变化,而其他微生物的水平却显著更高。经典的分析方法检测到水平升高的微生物的相对丰度较高,但它也报告指出该特定微生物的水平降低了,尽管实际上并未改变,从而导致了假阳性结果。这是因为所有微生物的相对丰度之和为100%。当某些微生物的相对丰度显著增加时,其他微生物的相对丰度必然会降低,这不能准确反映相对于宿主根组织的定殖微生物数量(图1b,c)。因此,经典分析方法的主要问题是它不能揭示相对于宿主植物根组织量的微生物总量,从而产生不准确的结果。
迄今,已经有多种方法来评估微生物组中相对于宿主组织的微生物总量被报道。在人类肠道中,将16S核糖体RNA(rRNA)测序与基于流式细胞仪的微生物细胞计数或微生物定量PCR(qPCR)结合使用,以定量粪便样品中的绝对微生物丰度。但是,这两种方法都不适合研究与宿主植物组织(如根相关的微生物组)相关的微生物总量。流式细胞术不能精确地计数活的或破坏的根样品中的微生物和植物细胞,通用的16S rRNA或ITS引物通常无法区分微生物序列与植物质体序列(叶绿体和线粒体),这妨碍了qPCR准确检测微生物DNA 。此外,诸如Card-FISH之类的染色方法也不适合以高通量方式检测和分类与植物根际的单个微生物组成员。还有研究提出了用植物细胞质体DNA作为解决方案,但由于每个细胞质体的数量高且拷贝数可变,因此它不是理想的内参。质体序列通常在根微生物组数据中占16S rRNA序列的80%以上,并在文库制备方案中通过实验去除。相比之下,基于宏基因组的测序可通过将读段分类为微生物和植物来检测植物组织中的根部微生物总量,但由于成本高昂,因此无法应用于大量样品。
内参标定追踪的利用是解决量化微生物总量的前瞻性策略。在RNA测序、蛋白质组学和宏基因组学领域已经应用了人工合成内参。最近,已开发出在微生物群落中不存在的已知序列或培养菌,或使用16S人工合成内参以及16S、18S和ITS人工合成内参的组合,用于定量微生物组分析。但是,这些基于spike-in的微生物组分析方法仅用于没有宿主的微生物组样品。另外,诸如16S rRNA基因或ITS的通用微生物基因与植物基因组中的序列具有高度的序列相似性,使得不可能通过比较微生物和植物中的基因来检测微生物组总量。因此,定量检测宿主组织如植物根部的微生物组丰度仍然是主要挑战。在这项研究中,我们开发了一种简单,经济高效且模块化的宿主相关定量丰度分析(HA-QAP)方法,该方法可通过以下方法检测相对于宿主植物的微生物总量和单个根系微生物组成员的定殖量,我们用微生物标记基因(16S rRNA基因或ITS)的拷贝数相对植物基因组的比例来衡量。这种高通量技术可以检测单个微生物的定殖,并准确评估与根宿主-微生物组以及微生物-微生物之间的相互作用。使用模拟实验,扰动实验和宏基因组测序,我们验证了HA-QAP的准确性和可重复性。此外,使用HA-QAP,我们发现微生物总量的增加是水稻植株干旱胁迫下微生物组和小麦根腐病中微生物组发生变化的关键特征。
图1.植物根部微生物组定量丰度分析的优势和实验步骤
Figure 1. Advantages and Experimental Procedure for Quantitative Abundance Profiling of the Microbiome in Plant Roots.
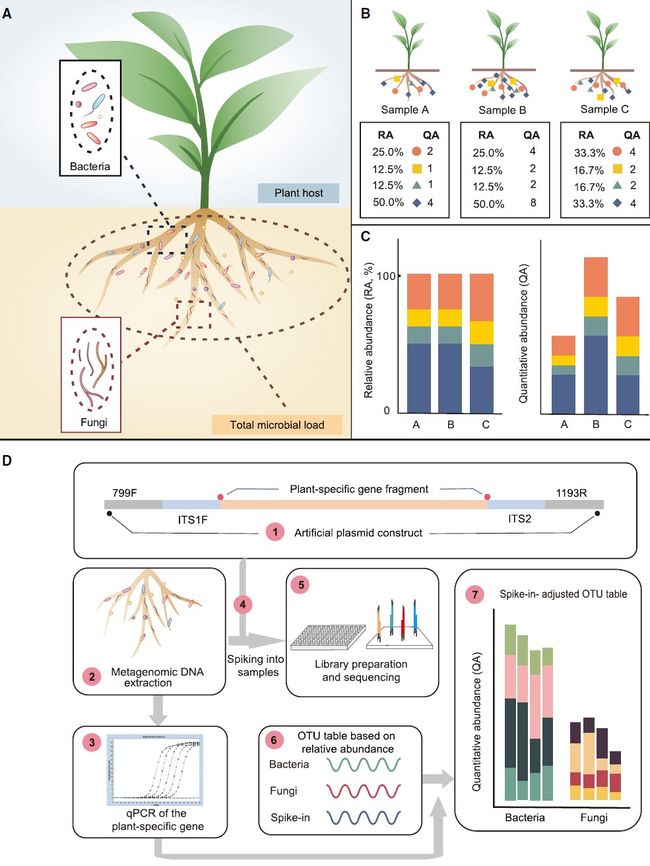
(A)当前基于相对丰度的根微生物组谱分析技术的主要局限性在于缺乏一种定量评估每单位植物根组织中微生物总量的方法。
(B)有着相同数量根组织的三种模拟的根微生物群(上图)和基于相对或定量丰度的相应微生物谱(下图)。 RA,相对丰度;QA,定量丰度,代表微生物标记基因相对于植物基因组的拷贝数比例。
(C)条形图显示了(B)中模拟的根微生物组样品的相对微生物分布图(左图)与定量微生物分布图(右图)的比较。
(D)示意图,显示了HA-QAP的工作流程。 (1)设计并构建人工质粒作为内参对照,包含保守的16S rRNA,ITS引物区(799F / 1193R和ITS1F / ITS2)和植物特异性基因序列(本研究中的RID1基因) 。 (2)从包括植物和微生物在内的根微生物组中提取DNA。 (3)通过qPCR对总DNA中发现的所选植物标记基因进行定量;该值用于在步骤(7)中将微生物相对丰度校准为定量丰度。 (4)根据经验,将预定量的内参质粒添加到根微生物组样品的DNA中。 (5和6)制备PCR扩增子文库(分别为细菌和真菌),测序(5)和分析(6)。 (7)基于内参的读长(reads)计数和在步骤(3)中通过qPCR定量的植物标志物基因的量来校准细菌和真菌的读长,以确定相对于宿主植物组织的定量丰度。
结果
spike-in内参质粒和HA-QAP方法的原理
The Spike-In Plasmid and Principle of HA-QAP
首先,我们合成了一个人工spike-in质粒作为内参,以定量检测相对于宿主植物的根部微生物组的总量和丰度谱。spike-in质粒包含植物特异性基因片段,其侧翼是细菌16S rRNA基因和真菌ITS的保守区(方法)。植物基因片段是RID1基因,编码水稻中开花的主要调控因子,而微生物基因组中不存在该调控基因。该基因片段可以被扩增并整合到经典的基于PCR的细菌和真菌谱中(图1d)。由于自然微生物群落中不存在扩增的植物特异性序列,因此可以在数据分析过程中轻松识别spike-in序列。在PCR扩增之前,我们向根部微生物组样品的DNA(包含植物和微生物DNA)中添加了一定量的spike-in质粒,并使用植物标记基因(例如水稻基因组中的RID1)通过qPCR测量了植物DNA ,并确定spike-in量与植物DNA之间的关系(详见方法)。然后基于这种关系,可以将微生物数据标准化为相同量的植物DNA,这反映了宿主植物根部定量的微生物定殖(图1d和方法)。
内参质粒(spike-in)适用于定量分析微生物组谱
The Spike-In Plasmid Is Suitable for Quantitative Microbiome Profiling
由于在分析结果中spike-in序列reads数与DNA模板中spike-in质粒的数量之间的相关性对于基于spike-in的定量至关重要,因此我们使用不同量的 spike-in质粒测试了这种相关性。如图2a所示,我们发现,遵循Poisson广义线性回归模型(Pearson’s r = 0.94,P<0.001),测序数据中 spike-in质粒的reads数随着输入 spike-in数量的增加而增加。该结果表明,在每个反应4.0×104至8.0×105个拷贝的动态范围内,对spike-in质粒的定量检测是可靠的,在这种动态范围内,我们可以预期 spike-in的read数相对于输入量线性变化(见补充表格1)。
为了确定 spike-in质粒是否影响微生物相对丰度的定量分析,我们spike-in质粒进行了梯度浓度探究(spike-in质粒浓度分别为0,4.0×104,8.0×104,1.6×105和8.0×105拷贝/PCR),使用细菌模拟实验中的DNA确定了单个细菌的相对丰度。然后,我们在去除spike-in序列后确定了了每个参考细菌菌株的相对丰度。含有 spike-in质粒的样品中单个细菌菌株的相对丰度与没有 spike-in质粒的对照样品的相对丰度保持一致(图2b; Wilcoxon秩和检验,P > 0.05,补充表2)。我们的结果表明,spike-in质粒不影响细菌PCR扩增和检测,并表明 spike-in质粒适用于定量微生物组谱分析。
图2. 在细菌模拟实验中HA-QAP比传统的基于相对丰度的分析更准确
Figure 2. HA-QAP Is More Accurate than Classical Profiling Based on Relative Abundance in the Bacterial Mock Experiments
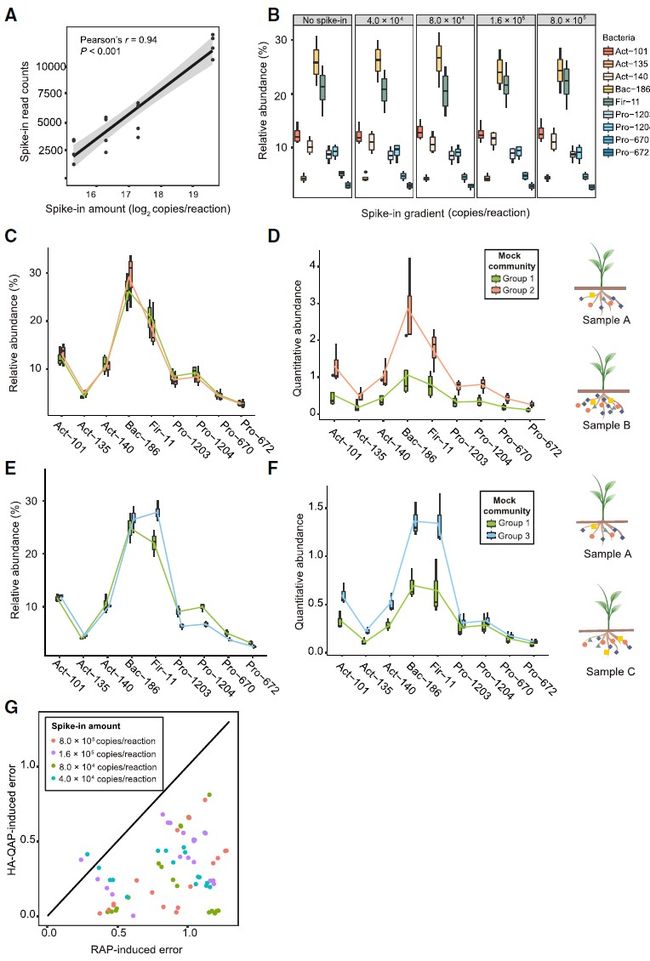
(A)Illumina测序获得的内参质粒(BI12-4)的reads数与DNA样品中内参质粒的数量之间线性相关的剂量反应曲线,表明测序数据中内参质粒reads反映了初始DNA样品中内参质粒的数量。灰色区域表示95%置信区间(CIs)。
(B)表示同一模拟实验中9种细菌的相对丰度的箱线图,每个反应的内参水平梯度为0-8.0×105份。 Wilcoxon秩和检验表明,没有内参的对照组和内参水平不同的组(每个反应4.0×104、8.0×104、1.6×105和8.0×105拷贝)之间的细菌相对丰度没有显著差异, P > 0.05)。
(C和D)HA-QAP方法显示细菌载量显著增加(双尾t检验,P < 0.05),这是基于相对丰度的经典方法无法检测到的。箱线图显示了使用基于相对丰度(RAP)(C)和HA-QAP(D)的经典方法在第1组和第2组之间进行模拟实验时细菌概况的比较。定量丰度代表细菌16S rRNA基因相对于植物基因组的拷贝数比。
(E和F)HA-QAP方法改进了对放线菌,拟杆菌和厚壁菌水平升高的检测(双尾t检验,P <0.05),并且揭示了变形杆菌的定量丰度没有变化(双尾t检验,P> 0.05)当其他细菌的数量增加时。箱线图显示了在模拟实验中使用RAP(E)和HA-QAP(F)在第1组和第3组之间进行细菌比较的情况。定量丰度代表细菌16S rRNA基因相对于植物基因组的拷贝数比。值得注意的是,RAP方法显示了变形菌门假阳性的降低,但HA-QAP却没有。
(G)散点图,显示了HA-QAP和RAP之间的误差比率。固定斜率= 1的线上的点表示HA-QAP产生的误差与RAP产生的误差相等。大多数点低于斜率= 1的线,表明HA-QAP方法可显示更多真实数据。数据基于不同内参水平下的比较(第1组与第2组;第1组与第3组)。设计了三组模拟实验。对于组1、2和3,n = 3、4和5。
(C)至(F)中显示的所有数据均来自每个反应带有4.0×104 拷贝的spike-in样品。使用其他浓度的spike-in,趋势是一致的。
Act, Actinobacteria; Bac, Bacteroidetes; Fir, Firmicutes; Pro, Proteobacteria
在人工混菌实验中检测微生物总量以评估方法的设计和合理性
Design and Rationale to Detect Microbial Load in Mock Experiments
为了验证HA-QAP的可靠性,我们将该新方法与基于相对丰度分析(RAP)的经典方法进行了比较,该方法使用了三组来自培养微生物的预混合DNA(模拟实验,图2c-f)。这三组模拟样本(如图1b所示)代表了使用RAP方法产生假阴性和假阳性结果的典型情况。第一组含有来自无菌植物,九种细菌和三种真菌的固定比例的混合DNA,其中包括四种细菌(放线菌,拟杆菌,厚壁菌和变形杆菌)和两种真菌(子囊菌和担子菌)的菌株;第2组含有与第1组等量的无菌植物DNA,但具有相同种间比例的微生物DNA为2倍;第3组含有与第1组等量的无菌植物,变形杆菌和子囊菌DNA,但来自具有相同种间比例的其余微生物的微生物DNA量是其两倍(补充表3)。为了确定用于正确分析的适当 spike-in浓度,我们使用 spike-in质粒(0.4.0×104,8.0×104,1.6×105和8.0×105拷贝/PCR)的梯度对每个组进行了定量分析。由于我们知道模拟实验中第1组和第2组之间以及第1组和第3组之间的确切差异,我们分别评估了基于 spike-in的HA-QAP和经典RAP方法的准确性。
HA-QAP在模拟实验中检测根微生物组的微生物总量
HA-QAP Detects the Microbial Load of Root Microbiome in Mock Experiments
在模拟实验中,HA-QAP方法检测到总细菌载量相对于植物根部DNA的变化(图2c,d)。由于经典的16S分析只能检测到微生物组中细菌成员的相对丰度,因此无法反映相对于植物根部的细菌总量的变化,这在样品之间进行比较时会产生假阴性结果(样品中的A和B图1b,c)。如上所述,我们比较了第1组和第2组中HA-QAP和RAP方法的准确性。经典的RAP方法揭示了第1组和第2组中每个菌株的相对丰度相同(双尾学生t检验,P > 0.05),这没有反映相对于根的细菌总量和丰度(图2c;补充表4)。相比之下,HA-QAP方法在第1组和第2组之间检测到的细菌丰度相对于植物根部DNA的平均变化为2.4倍(预期倍数为2倍),每次PCR的 spike-in浓度为4.0×104拷贝(双尾t检验,P < 0.05;图2d;补充表4)。在其他三个实验中也观察到了相同的趋势,不同数量的 spike-in质粒(每个PCR分别为8.0×104,1.6×105和8.0×105拷贝/PCR)(补充图1;补充表4)
HA-QAP可在模拟实验中准确检测单个根微生物组成员的变化
HA-QAP Accurately Detects the Changes of Individual Root Microbiome Members in Mock Experiments
当模拟实验中高丰度的细菌发生显著变化时,HA-QAP方法检测到相对于根DNA的单个细菌的定量丰度(图2e,f)。当高丰度的细菌增加或减少时,经典的RAP方法(基于相对丰度)通常会产生假阳性结果。我们使用HA-QAP和RAP方法在两个模拟实验(第1组和第3组)中研究了微生物群落。经典的RAP方法在第1组至第3组检测到变形菌(Pro-1203,Pro-1204和Pro-670)下降了(假阳性)(双尾Student t检验,P < 0.05)。它们相对于植物根的丰度没有变化(图2e;补充表5)。值得注意的是,HA-QAP方法检测到第1至3组细菌群落的真实变化,并且放线菌,拟杆菌和厚壁菌的丰度增加(双尾Student t检验,P < 0.05;平均倍数变化:1.9),当每次PCR的 spike-in浓度为4.0×104拷贝时,变形杆菌菌株的水平没有显著差异(双尾Student t检验,P > 0.05;图2f,补充表5)。在使用其他三种 spike-in质粒梯度的实验中也观察到了这些趋势(补充图2;补充表5和6)。
根据模拟实验的结果,HA-QAP方法比RAP方法更准确。我们使用两种方法在微生物模拟实验中计算了期望值和测量值之间的误差(图2g)。我们绘制了HA-QAP误差与RAP误差之间的比率(图2g),其中斜率为1的线表示HA-QAP引起的误差与RAP引起的误差相等。 HA-QAP引起的误差和RAP引起的误差之间的比率大多数都在该线以下,这表明HA-QAP方法比RAP方法更准确。总之,HA-QAP方法克服了RAP方法的局限性,揭示了相对于根DNA的细菌总量变化和单个细菌成员的定量丰度。
与细菌模拟实验中观察到的结果相似,HA-QAP还检测了相对于植物根部的单个真菌菌株的微生物总量和定量丰度。 spike-in reads和输入量遵循Poisson通用线性回归模型(Pearson r = 0.98,P <0.001,图3a),表明 spike-in质粒的定量检测在每个反应4.0×104至8.0×106拷贝的动态范围内是可靠的(补充表7)。如图3b中所示, spike-in质粒不影响每种真菌的相对丰度的趋势(Wilcoxon秩和检验,P > 0.05,补充表8)。不出所料,经典的RAP方法未检测到第1组和第2组之间的总真菌总量差异(双尾Student t检验,P > 0.05;图3c,补充图3a;补充表9),而HA-QAP揭示了相对于第1组和第2组植物根DNA而言,真菌总量增加了(双尾Student t检验,P < 0.05;图3d,补充图3b;补充表9)。此外,基于 spike-in的HA-QAP方法检测到Basi-AF78真菌相对于植物根系的丰度有特定的增加(双尾Student t检验,P < 0.05;图3e,f)。相比之下,在RAP方法中,这种增加导致两种子囊菌分离物(Asco-AF1和Asco-AF105)的丰度假阳性降低(双尾Student t检验,P < 0.05;图3e),这在HA-QAP方法中是可以排除的(两尾Student t检验,P > 0.05;图3e,f;补充图4a,b;补充表10)。最后,我们计算了HA-QAP引起的误差与RAP引起的误差之间的比率,并绘制了值(图3g)。对于真菌模拟实验,所有点均以斜率 = 1下降到线下,这表明,正如对细菌所观察到的,HA-QAP方法比RAP方法更准确。
图3. 在真菌模拟实验中HA-QAP比基于相对丰度的经典分析更为准确
Figure 3. HA-QAP Is More Accurate than Classical Profiling Based on Relative Abundance in the Fungal Mock Experiments
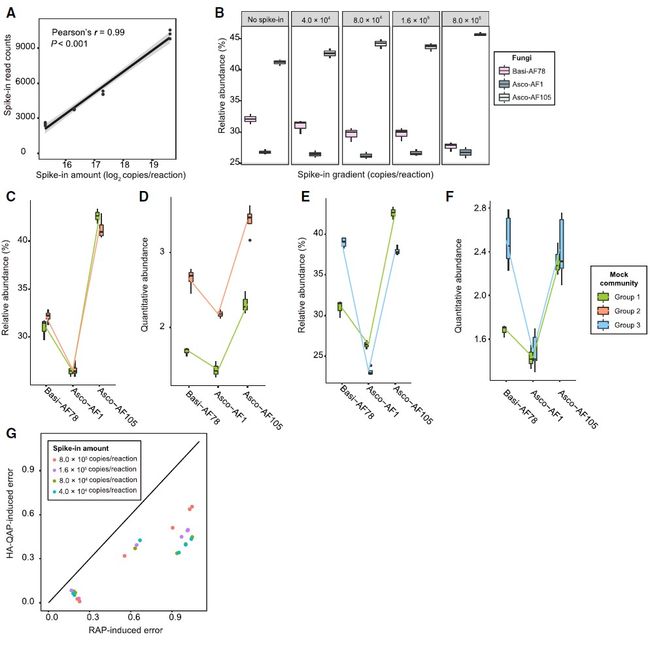
(A)通过Illumina测序获得的内参质粒(BI12-4)的reads数与DNA样品中内参质粒的数量之间线性相关的剂量反应曲线,表明测序数据中内参质粒reads反映了初始DNA样品中内参质粒的数量。灰色区域表示95%CI。
(B)箱线图代表在同一模拟实验中分配给三种真菌的reads的相对丰度,每个反应的内参水平梯度为0至8.0×105个拷贝。 Wilcoxon秩和检验显示,没有内参的对照组和内参水平不同的组(每个反应4.0×104、8.0×104、1.6×105和8.0×105拷贝)之间的真菌相对丰度没有显著差异, P > 0.05)。
(C和D)HA-QAP方法显示出真菌载量显著增加(双尾t检验,P <0.05),这是基于相对丰度的经典方法无法检测到的。箱线图显示了在模拟实验中使用RAP(C)和HA-QAP(D)在第1组和第2组样品之间的真菌谱的比较。定量丰度代表真菌ITS与植物基因组的拷贝数比例。
(E和F)HA-QAP方法更准确地检测到了担子菌水平的升高(双尾t检验,P <0.05),并揭示了子囊菌的定量丰度没有变化(双尾t检验,P > 0.05),其他真菌分离株的水平增加。箱线图显示了在模拟实验中使用RAP(E)和HA-QAP(F)在第1组和第3组之间对真菌谱的比较。定量丰度代表真菌ITS与植物基因组的拷贝数比。值得注意的是,RAP表现出了子囊菌水平的假性降低(Asco-AF1和Asco-AF105),而HA-QAP却没有。
(G)散点图,显示了HA-QAP和RAP之间的误差比率。固定斜率 = 1的线上的点表示HA-QAP产生的误差与RAP产生的误差相等。所有点均落在斜率 = 1的线以下,表明HA-QAP方法可显示更多真实数据。数据基于不同内参水平下的比较(第1组与第2组;第1组与第3组)。设计了三组模拟实验。对于组1、2和3,n = 4、3和5。
(C)至(F)中显示的所有数据均来自每个反应带有4.0×104份spike-in的样品。当使用其他峰值水平时,趋势是一致的。
Asco, Ascomycota; Basi, Basidiomycota
HA-QAP方法可检测自然根样品中微生物总量的变化
HA-QAP Detects Changes in Microbial Load in Natural Root Samples
由于HA-QAP方法在模拟实验中效果很好,因此我们进一步验证了该内参质粒在自然根样品中的效用。我们发现 spike-in质粒不会改变自然根样品中细菌或真菌的相对丰度。具有和不具有 spike-in质粒的自然根样品之间细菌和真菌操作分类单位(OTU)的相对丰度具有高度可比性(对于细菌,Pearson’s r = 0.99,图4a;对于真菌,Pearson’s r = 0.97,图4b;其他 spike-in浓度见补充图5)。香农指数在对照样品和 spike-in样品之间也没有显著差异(Wilcoxon秩和检验,P > 0.05,补充图6)。
为了测试HA-QAP方法在自然根样品中的性能,我们用来自无菌水稻根部的一定量的DNA扰动了自然根的DNA,人为地将样品的原始微生物总量降低至原始的自然样本的55%。 HA-QAP方法相对于受干扰样品中的植物根部组织检测到细菌和真菌的微生物总量降低(图4d,f),而经典RAP方法未检测到(图4c,e),因为经典RAP方法仅检测到微生物的相对丰度。 HA-QAP方法显示每个反应的 spike-in浓度为2.0×105拷贝时,细菌和真菌的总量分别降低了50%和55%(图4d,f)。在其他实验中,使用不同浓度的 spike-in质粒也观察到了类似的趋势(补充图7)。综上所述,HA-QAP方法成功地检测了模拟样品和自然样品中相对于根组织的微生物总量,因此可用于研究微生物组与根之间的定量相互作用。
图4. HA-QAP揭示了自然根样品中微生物总量的降低
Figure 4. HA-QAP Reveals a Reduction in Microbial Load in Perturbed Natural Root Samples
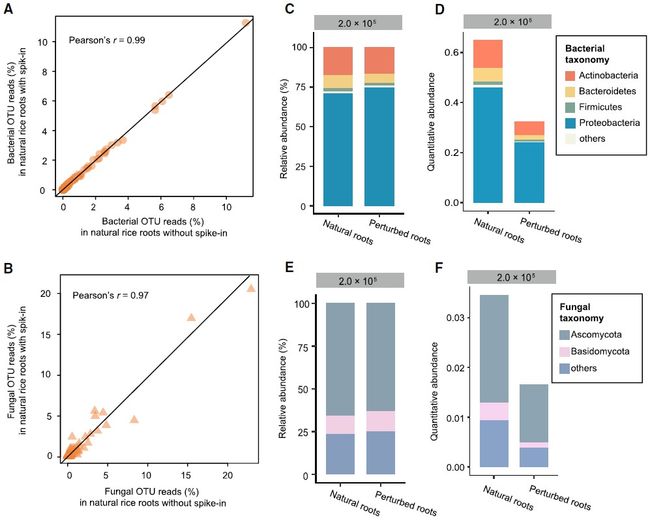
(A和B)散点图显示y轴上具有spike-in(每个反应2.0×105拷贝)的自然稻根中细菌OTU(A)和真菌OTU(B)的相对丰度之间的相关性)与没有内参(x轴)的水稻根部比较,表明内参不会影响自然根样品中细菌或真菌OTU的相对丰度。
(C和D)基于相对丰度(C)和定量丰度(D)的自然稻根和微生物含量为55%的稻根的细菌谱,分别与RAP和HA-QAP原始自然样品相比。定量丰度代表细菌16S rRNA基因相对于植物基因组的拷贝数比。请注意,HA-QAP方法揭示了被扰动的自然稻根中细菌含量的降低。
(E和F)基于相对丰度(E)和定量丰度(F)的真菌谱,分别基于RAP和HA-QAP与原始自然样品相比微生物含量为55%的微生物根量。定量丰度代表真菌ITS与植物基因组的拷贝数比。注意,HA-QAP方法揭示了被扰动的的自然稻根中真菌总量的减少。
(C)至(F)中的数据代表三个技术重复
微生物总量是水稻根系微生物组对干旱胁迫响应变化的关键特征
Microbial Load Is a Key Feature of the Changes in Root Microbiome Response to Drought Stress in Rice
干旱压力是阻碍农业生产力的最重要因素之一,每年造成全球数十亿美元的损失。确保全球粮食安全的努力越来越侧重于研究干旱条件下农作物中的根部微生物组。干旱显著改变了包括小麦,大麦,高粱和水稻在内的几种植物的根系微生物组组成。但是,干旱胁迫下根系微生物总量的变化是未知的。为了解决这个问题,我们在干旱和潮湿的条件下,在中国的海南农场和安徽农场种植了两个水稻品种MH63和WYJ7。
与先前的报道一致,我们发现基于微生物相对丰度干旱处理影响了与水稻根系相关的微生物组成(补充图8)。使用HA-QAP方法,我们发现海南农场在干旱条件下,水稻根系微生物组的细菌和真菌总量显著增加(图5a)。在干旱条件下,MH63和WYJ7中根系微生物组相对于宿主植物组织的总微生物总量分别增加了1.1倍和2.8倍(Wilcoxon秩和检验,P < 0.05,图5a)。为了验证通过HA-QAP方法检测到的干旱胁迫下植物根系中微生物总量的增加,我们对相同样品进行了宏基因组测序,并通过评估可比对到NCBI-NR数据库的微生物和植物中的reads的比例来评估微生物总量。宏基因组数据表明,微生物总量增加与通过HA-QAP方法检测到的微生物总量相关(图5c)。在干旱胁迫条件下,MH63和WYJ7的细菌总量分别增加了1.4倍和2.1倍(图5a)。此外,在距海南农场1650公里的安徽农场也发现了干旱胁迫下MH63和WYJ7品种根系微生物总量的增加趋势(补充图9a)。因此,我们的数据表明干旱介导的植物根系微生物总量的增加代表了植物微生物组对干旱胁迫的响应的内在特征。
为了评估微生物总量对干旱和潮湿条件下种植的水稻根际微生物组差异分类群模式的影响,我们比较了基于RAP和HA-QAP方法中富集(enriched)和下降(depleted)的特征。我们发现,在干旱条件下使用RAP方法检测到的水稻根系中富集的种群门水平和OTU水平,使用HA-QAP显示更加富集。相比之下,当使用HA-QAP时,使用RAP方法在干旱胁迫下减少的物种显示不那么明显,这表明经典RAP方法人为地减少了这些类群的相对丰度。在不同的水稻品种和不同的地点(海南和安徽农场;图5d,e;补充图9b-c)中都观察到了这些趋势。例如,在干旱条件下,RAP方法显示MH63根的放线菌水平没有显著增加,而HA-QAP方法相对于水稻根显示出增加了0.2倍。
在OTU水平上进一步说明了HA-QAP对差异细菌的影响。如图5f所示,仅当使用HA-QAP方法时,OTU_16(放线菌)才响应干旱而富集。对于通过RAP检测到的干旱条件下的OTU下调,HA-QAP分析的差异不显著或截然相反(图5g,h)。当通过RAP检验而不是HA-QAP检验时,OTU_11(Proteobacteria)对干旱的相应显著减少(图5f)。使用RAP,OTU_13(变形杆菌)在干旱胁迫的根样品中显著富集,而使用HA-QAP则是显著下调减少(图5g)。从RAP与HA-QAP方法获得的上述不同结果依赖于对植物组织中微生物总量的准确评估。 HA-QAP方法是一种更准确的工具,可用于识别潜在的有益农业的重要微生物,从而提高水稻的耐旱性。
图5. HA-QAP显示干旱胁迫下水稻根系的微生物总量增加
Figure 5. HA-QAP Reveals Microbial Load Increase in Rice Roots under Drought Stress
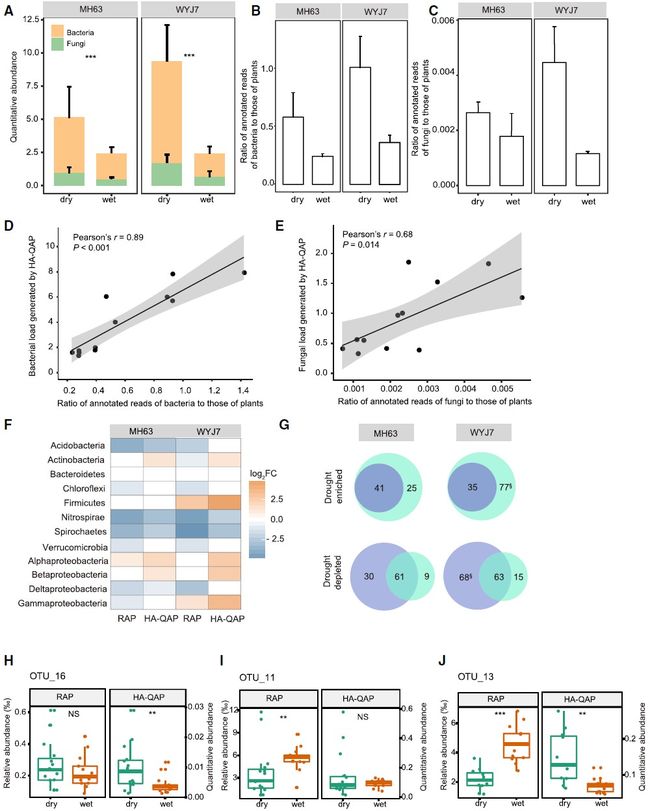
(A)HA-QAP方法显示,在干旱胁迫条件下,两个水稻品种(MH63和WYJ7)的根微生物组中细菌和真菌的载量显著增加。定量丰度代表细菌16S rRNA基因和真菌ITS相对于植物基因组的拷贝数比。
(B和C)宏基因组数据显示,在干旱条件下,海南种植的水稻MH63和WYJ7品种的带注释的细菌(B)和真菌(C)的reads相对于宿主植物的比率更高(干:n = 3;湿:n = 3)。
(D和E)散点图显示注释的细菌(D)和真菌(E)reads与通过宏基因组测序(x轴)检测到的植物的注释reads的比率与通过HA-QAP方法检测到的微生物总量之间的线性相关性(y轴)。灰色区域表示95%CI。
(F)热图显示了通过HA-QAP和RAP检测到的水稻品种MH63和WYJ7中干旱响应细菌门的丰度相对于对照处理(湿)的log2倍变化。橙色框表示增加,而蓝色框表示减少。白框表示没有明显的倍数变化。请注意,对于两个品种的根部微生物组,HA-QAP方法均显示出干旱响应门的变化,与湿条件相比,干旱胁迫条件下富集的富集度提高且下调减少,这是由于通过HA-QAP检测到的根细菌总量增加所致。
(G)Venn图显示了通过RAP和HA-QAP方法检测到的干旱响应型OTU的重叠和差异。紫色和绿色椭圆分别表示通过RAP和HA-QAP方法检测到的干旱响应型OTU。 § 表示使用RAP被检测为减少下调的OTU但使用HA-QAP被鉴定为富集的五个OTU,这表明微生物总量值对于跟踪这些细菌的丰度变化至关重要。
(H–J)示例显示了使用RAP和HA-QAP在干旱和潮湿条件下水稻根中OTU的变化。定量丰度代表细菌16S rRNA基因相对于植物基因组的拷贝数比。三种OTU对干旱胁迫条件的响应因方法而异:仅使用HA-QAP将OTU_16(H)检测为对干旱有响应的富集。相反,仅使用RAP而非HA-QAP,在干根样品中OTU_11(I)被检测到显著减少。发现使用RAP在干根样品中OTU_13(J)明显减少,但使用HA-QAP富集。
注意,海南的所有数据与安徽的数据一致(另见补充图9和10)。对于所有分析(B和C除外),生物学重复次数如下:MH63(干:n = 15;湿:n = 13); WYJ7(干:n = 11,湿:n = 13);大块土壤(干:n = 6;湿:n = 6)。误差棒 代表根据重复计算得出的SD。通过Wilcoxon秩和检验确定统计学显著性。星号表示统计学上的显著差异(** P < 0.01; *** P < 0.001)。 NS,不显著
HA-QAP显示感染根腐病的小麦根际微生物总量绝对增加
HA-QAP Reveals Absolute Increases in Total MicrobialnLoad in Wheat Roots Infected with Root Rot Disease
普通根腐病是一种冬小麦疾病,每年导致产量损失。症状包括根冠下节间和茎基部的棕褐色病变。关于根腐病对小麦根系微生物组组成和微生物总量的影响知之甚少。我们使用HA-QAP方法研究了健康小麦植物和在田间条件下表现出常见根腐病的小麦植物中的根微生物组。我们发现,与健康植物相比,患有根腐病的植物显示出不同的根微生物组组成并增加了微生物总量。首先,基于微生物组成的主坐标分析(PCoA)显示,健康和受感染的小麦植物的根微生物群在第一坐标和第二坐标中分开(图6A和6B)。对于细菌微生物组,在PCoA2坐标上观察到以健康状况区分的根样品清晰分离,同时解释了22.4%的差异(PERMANOVA,P < 0.001)。对于真菌群落,健康样本和感染样本在前两个坐标轴上明显分开,解释了总变异的23.2%(PERMANOVA,P < 0.001)。其次,HA-QAP方法显示感染样品中根部微生物组的细菌和真菌总量显著增加(Wilcoxon秩和检验,P <0.05,图6C)。根腐病样品中的总细菌总量增加了2.2倍,而真菌总量增加了4.5倍。值得注意的是,考虑到总微生物总量(Wilcoxon秩和检验,P < 0.005),相对于植物根部,潜在疾病相关Bipolaris的丰富度显著增加(而使用RAP方法时,重要的微生物信号却丢失了(P > 0.05)(图6D)。宏基因组测序进一步验证了细菌和真菌总量的增加(图6E)。因此,尽管RAP允许我们区分健康的小麦植物和受感染的小麦植物,但只有HA-QAP分析才能确定增加的微生物丰度是与常见根腐病相关的微生物组变化的关键特征。微生物不是孤立存在的,而是形成复杂的生态相互作用的。共生网络被广泛用于通过微生物相对丰度的相关性推断微生物相互作用。然而,此类分析的结果应谨慎解释,因为它们容易受到基于相对丰度的组成的影响。为了评估微生物总量对共丰度模式的影响,我们使用RAP和HA-QAP方法,根据丰度前55个属和Bipolaris潜在病菌的丰度信息,构建了一个共现网络。如图6F所示,与RAP相比,HAQAP方法(上三角)检测到更多的共变(co-varying)属,包括与Bipolaris相关的16个属(细菌,13;真菌,3; P < 0.05)。与HA-QAP方法相反,RAP方法(下部三角形)仅检测到三个与Bipolaris相关的属。综上所述,与RAP方法相比,HA-QAP方法识别出的微生物总量增加所驱动的更多潜在的疾病相关微生物组信号,这可能有助于研究常见根腐病中的微生物与微生物相互作用以及与微生物相关的发病机理。
图6. HA-QAP显示出患有根腐病的小麦植株中微生物总量增加
Figure 6. HA-QAP Reveals Microbial Load Increase in Wheat Plant with Root Rot Disease
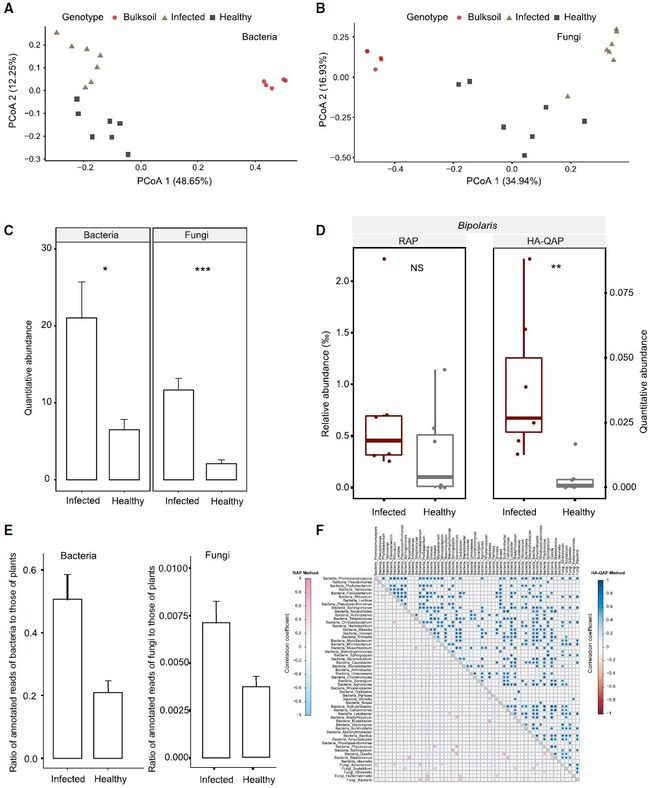
(A和B)Bray-Curtis距离的主坐标分析表明,感染的根和健康的根在细菌(A)或真菌(B)微生物组中形成了不同的聚簇。每个点对应一个样本:受感染的根样本,三角形;健康的根样品,正方形;散装土壤样品,圆圈。
(C)感染的根样品具有比健康根显著更高的细菌和真菌载量。定量丰度代表细菌16S rRNA基因和真菌ITS相对于植物基因组的拷贝数比。
(D)通过RAP和HA-QAP评估的健康和感染根腐病样品中潜在疾病相关致病菌双极属的相对数量和相对定量数量。定量丰度代表真菌ITS与植物基因组的拷贝数比。
(E)宏基因组数据显示,感染根样品(感染样品,n = 4;健康样品,n = 4)中细菌(左)和真菌(右)reads与宿主植物reads的比率更高。
(F)通过RAP和HA-QAP检测到的前55个最丰富属中的属共生模式以及潜在的与疾病相关的致病性双极属。使用RAP(下三角)和HA-QAP(上三角)计算分类单元丰度之间的成对相关性。显著相关性(双属检验校正,fdr <0 .05)用圆圈表示;每个圆圈的颜色代表相关系数(spearman ρ)。首先根据细菌和真菌域对分类单元进行排名,然后根据同一域中个体的定量丰度对其进行排序。最左边/最上面代表丰度最高。
对于所有分析(E除外),生物学重复次数如下:感染的根样品,n = 7;健康的根样品,n = 7;散装土壤样品,n =5。Error bars表示根据重复计算得出的SD。通过Wilcoxon秩和检验确定统计学显著性。星号表示统计学上的显著差异(* P < 0.05; ** P < 0.01)。 NS,不显著。
讨论
环境因素,营养成分和疾病会影响微生物对宿主的定殖。通过将spike-in DNA整合到根微生物组样品中,我们建立了一种简单,高通量的方法来检测根微生物组相对于宿主植物组织的总微生物总量。 HA-QAP这种方法通过预定义的 spike-in质粒和植物DNA的比例关系,将微生物组与宿主植物连接起来。我们通过模拟实验(图2、3),扰动实验(图4)和自然样品的宏基因组测序(图5b)验证了HA-QAP方法。通过在潮湿和干旱条件下将HA-QAP应用于水稻植株的根部微生物组,我们证明了干旱条件下两个地方种植的两个水稻品种的水稻根系微生物组的微生物总量增加(图5),表明这种变化微生物总量中的微生物含量是暴露于干旱胁迫下的水稻植株根系微生物组的关键特征。
由于HA-QAP方法基于微生物总量,因此可以揭示单个微生物相对于宿主植物组织的定量丰度,从而避免了从基于相对丰度(RAP)的经典方法获得的假阴性和假阳性结果。将HA-QAP方法应用于模拟实验,我们证明了当样品中相对微生物组成相同时,HA-QAP检测到单个微生物相对于根部组织的水平增加(RAP中的假阴性;图2c,d和图2c)。 3c,d)。由于单个微生物的定量丰度不受其他微生物的定量丰度的影响,因此HA-QAP方法避免了由于高丰度微生物水平的增加而导致未改变的微生物组成员变化的错误检测(RAP中的假阳性;图。 2e,f和3e,f)。因此,HA-QAP方法可提供有关单个微生物组成员方向性和范围变化的准确信息。
植物生活在非生物和生物胁迫条件下,这会严重影响根部微生物组。 HA-QAP使我们能够识别与植物根部相关的有益或有害微生物。例如,放线菌是在干旱条件下种群增加的最显著的门类,当我们使用HA-QAP而非RAP进行测量时,在我们的试验田中,对干旱的响应显著富集(图5)。除了非生物胁迫外,各种生物胁迫对与植物相关的微生物组也有深远的影响,据认为这代表了抑制疾病的微生物机制。这项研究中使用的 spike-in质粒使我们能够同时使用细菌和真菌引物组扩增DNA,以观察微生物组成员对宿主的协同作用。这些信息特别有用,为进一步研究植物-细菌-真菌的相互作用提供了指导。
鉴于其准确性和技术可行性,HA-QAP方法扩展了基于相对丰度的经典微生物组分析。该方法应有助于揭示微生物组在未来植物健康和生产中的作用的重要生物学机制。稍作修改,HA-QAP可用于宿主-微生物组相互作用的任何类型的扩增子图谱研究。
Reference
Xiaoxuan Guo, Xiaoning Zhang, Yuan Qin, Yong-Xin Liu, Jingying Zhang, Na Zhang, Kun Wu, Baoyuan Qu, Zishan He, Xin Wang, Xinjian Zhang, Stéphane Hacquard, Xiangdong Fu & Yang Bai. Host-Associated Quantitative Abundance Profiling Reveals the Microbial Load Variation of Root Microbiome. Plant Communications 1, 100003, doi:https://doi.org/10.1016/j.xplc.2019.100003 (2020).
撰文:张小宁 中科院遗传发育所
责编:刘永鑫 中科院遗传发育所
猜你喜欢
- 10000+: 菌群分析
宝宝与猫狗 提DNA发Nature 实验分析谁对结果影响大 Cell微生物专刊 肠道指挥大脑 - 系列教程:微生物组入门 Biostar 微生物组 宏基因组
- 专业技能:生信宝典 学术图表 高分文章 不可或缺的人
- 一文读懂:宏基因组 寄生虫益处 进化树
- 必备技能:提问 搜索 Endnote
- 文献阅读 热心肠 SemanticScholar Geenmedical
- 扩增子分析:图表解读 分析流程 统计绘图
- 16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
- 在线工具:16S预测培养基 生信绘图
- 科研经验:云笔记 云协作 公众号
- 编程模板: Shell R Perl
- 生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”


点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA