用R包TCGA-Assembler下载TCGA数据
今天师兄发给我一个TCGA网址,让我想办法把胰腺癌的所有RNA-Seq数据下载下来,可是界面是这样的:
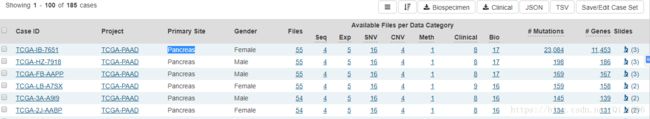
这么多样本有各自的测序数据,甲基化数据,突变数据和临床数据等,不可能一个个去点开下载,那么批量下载就可以借助R的一个包TCGA-Assembler。电脑是win10系统,所以以下教程是基于windows系统的。
安装教程:1.建立新文件夹,例如:E:\TCGA_assemble;然后下载TCGA-Assembler安装包(链接:https://pan.baidu.com/s/1WMxP-0ucJxGR7xwg743b6w 密码:33s2)并解压,将解压之后的文件夹里的Module_A.R和Module_B.R复制到刚才建立的文件夹内,同时将curl.exe这个文件,复制到电脑C盘Windows文件夹的System32这个文件夹中。
2.在TCGA_assemble文件夹内建立一个用来存放下载数据的新文件夹(注意一定要在TCGA_assemble文件夹内),比如我下载的数据是pancreatic adenocarcinoma,所以简写命名为PAAD。现在你的TCGA_assemble文件夹内应该是这样的:

3.接下来打开R,并把路径设置到TCGA_assemble文件夹的位置,然后导入刚才的两个模块,在导入模块的过程中可能会出现缺包的情乱,那当然是缺什么包装什么包,这个应该大家都会啦。因为是新电脑,所以要装的包比较多,代码如下:
#设置路径到刚才的文件夹
setwd("E:/TCGA_assemble")
#载入所需的包
source("http://bioconductor.org/biocLite.R")
biocLite("httr")
biocLite("RCurl")
biocLite("stringr")
biocLite("HGNChelper")
biocLite("rjson")
library(httr)
library(RCurl)
library(stringr)
library(HGNChelper)
library(rjson)
#载入TCGA_assemble文件夹中的两个模块,其中A模块用来下载数据,B用来分析数据
source("Module_A.R")
source("Module_B.R")
以上步骤完成后没有报错就可以开始下载数据了。
4.大家在下载前可以先打开解压后的文件夹内的指南,里面有下载每种类型数据的代码和说明。我觉得还是十分清晰明了的。

4.1首先给大家讲一下RNA-Seq数据,也就是大师兄给我的任务。
给大家看一下指南里关于RNA-Seq数据下载的代码是这样的:

第一个参数是cancerType,也就是你要弄清楚下载数据是什么癌症以及它的缩写是什么,具体可以查看网址 https://tcga-data.nci.nih.gov/docs/publications/tcga/。
第二个参数是你需要知道下载的数据是在什么平台测得的,自己去TCGA的网站看下数据信息,如下是部分截图

第三个参数则是组织类型,如果有就写上去,如果没有就默认全选。
第四个参数则是下载的数据保存的位置。
第五个参数则是下载的数据文件名字。
第六个参数则是TCGA上自己挑选的样本ID,一般形式为TCGA-XX-XXXX。
那我的是185个pancreatic adenocarcinoma(PAAD)的RNA-Seq数据,代码如下:
setwd("E:/TCGA_assemble/PAAD")
Patient_ID<-read.table(file="TCGA_ID.txt",sep=" ",quote="\t",header=F)
vPatient_ID<-as.vector(as.array(Patient_ID[,1]))
#A.下载数据
#下载RNA-Seq数据
#' choose a cancer type
#' 可查看网址https://tcga-data.nci.nih.gov/docs/publications/tcga/
filename_READ_RNASeq <- DownloadRNASeqData(cancerType = "PAAD",
assayPlatform = "gene.normalized_RNAseq",
saveFolderName = "./gene_RNA",inputPatientIDs = vPatient_ID)
需要说明的是一般inputPatientIDs=c("TCGA-05-4245", "TCGA-44-2661", "TCGA-38-4630", "TCGA-93-8067", "TCGA-05-5420")是这样的,但是样本太多,所以就想到了比较笨的方法,在网页上将所有样本选中,点击右上角TSV将当前信息下载下来获取Case ID,也就是PatientID,再导入R进行转换。

运行后如果出现如下界面则表明数据下载完成:
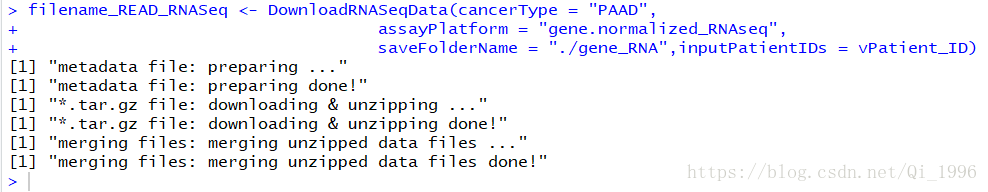
最后下载的数据就是这样的(截图比较粗糙,捂脸):


4.2下载甲基化数据同上,代码如下:
#下载甲基化数据
filename_READ_Methylation450<- DownloadMethylationData(cancerType = "PAAD",
assayPlatform = "methylation_450", saveFolderName = "./MethylationData",
inputPatientIDs =vPatient_ID)
其他类型数据同样如此,该模块提供了下载这么多类型的数据方法,大家可以根据需要去修改代码,不过一定要搞清楚癌症类型,数据平台和PatientID,

还有模块B是用来分析数据,不过我还米有做,等之后做了就会来更新哒。新人一枚,如有错漏之处欢迎指正。