SCENIC是一种基于单细胞RNA-seq数据推断基因调控网络及其相关细胞状态的工具
这里介绍的步骤方法是针对在R环境中(GENIE3),主要是GENIE3/GRNBoost这两种方法的选择,数据量大的推荐使用GRNBoost(in Python),参考https://www.jianshu.com/p/eccfe2d1b2c7
SCENIC分析步骤:
建立基因调控网络
- 根据每个转录因子的共表达确定它的潜在靶点;
·对表达矩阵进行过滤+running GENIE3/GRNBoost
·将上一步骤中得到的targets构建共表达模型 - 根据DNA motif分析,挑选潜在的直接结合的靶点(regulons: transcription factors (TFs) and their target genes, together defining a regulon);
鉴定细胞状态及其调控元件
- 对每一个细胞分析网络活性;
·对细胞中的regulons进行打分
·可选步骤:将network activity转换成ON/OFF格式 - 根据基因调控网络活性鉴定稳定细胞状态+对结果进行进一步探索;
1. 输入文件格式:single-cell RNA-seq expression matrix
a) .loom文件
#.loom文件能够直接导入SCENIC
# 下载.loom文件: download.file("http://loom.linnarssonlab.org/clone/Previously%20Published/Cortex.loom", "Cortex.loom")
loomPath <- "Cortex.loom"
#导入数据
library(SCopeLoomR)
loom <- open_loom(loomPath, mode="r")
exprMat <- get_dgem(loom)
cellInfo <- get_cellAnnotation(loom)
close_loom(loom)
b) 10X/CellRanger下机数据
load the CellRanger output into R
c) 其他R对象(e.g. Seurat、SingleCellExperiment)
#for a SingleCellExperiment object
sce <- load_as_sce(loomPath) # any SingleCellExperiment object exprMat <- counts(sce)
cellInfo <- colData(sce)
#for a Seurat object
# 首先选择你要提取的细胞
cells.use <- WhichCells(object = immune.combined, ident = 3)
# 这里的assay指的是你要提取的表达矩阵类型
expr <- GetAssayData(object = immune.combined, assay= "RNA", slot = "data")[, cells.use]
# 转换格式
expr <- as(Class = 'matrix', object = expr)
cellInfo <- data.frame(seuratCluster=Idents(seuratObject))
d) GEO数据
# dir.create("SCENIC_MouseBrain"); setwd("SCENIC_MouseBrain") # if needed # (This may take a few minutes)
if (!requireNamespace("GEOquery", quietly = TRUE)) BiocManager::install("GEOquery")
library(GEOquery)
geoFile <- getGEOSuppFiles("GSE60361", makeDirectory=FALSE) gzFile <- grep("Expression", basename(rownames(geoFile)), value=TRUE)
txtFile <- gsub(".gz", "", gzFile)
gunzip(gzFile, destname=txtFile, remove=TRUE)
library(data.table)
geoData <- fread(txtFile, sep="\t")
geneNames <- unname(unlist(geoData[,1, with=FALSE]))
exprMatrix <- as.matrix(geoData[,-1, with=FALSE])
rm(geoData)
dim(exprMatrix)
rownames(exprMatrix) <- geneNames
exprMatrix <- exprMatrix[unique(rownames(exprMatrix)),] exprMatrix[1:5,1:4]
# Remove file downloaded:
file.remove(txtFile)
cellLabels <- paste(file.path(system.file('examples', package='AUCell')), "mouseBrain_cellLabels.tsv", sep="/")
cellLabels <- read.table(cellLabels, row.names=1, header=TRUE, sep="\t")
cellInfo <- as.data.frame(cellLabels)
colnames(cellInfo) <- "Class"
2. 初始化数据
2.1 数据导入
#创建空文件夹
dir.create("SCENIC_MouseBrain")
setwd("SCENIC_MouseBrain")
#导入数据,这里用到的是包自带数据
loomPath <- system.file(package="SCENIC", "examples/mouseBrain_toy.loom")
library(SCopeLoomR)
loom <- open_loom(loomPath, mode="r")
exprMat <- get_dgem(loom)
cellInfo <- get_cellAnnotation(loom)
close_loom(loom)
dim(exprMat)
## [1] 862 200
2.2 细胞聚类信息/表型数据
#对感兴趣的变量赋予特定的颜色
head(cellInfo)
## Class nGene nUMI
## 1772066100_D04 interneurons 170 509
## 1772063062_G01 oligodendrocytes 152 443
## 1772060224_F07 microglia 218 737
## 1772071035_G09 pyramidal CA1 265 1068
## 1772067066_E12 oligodendrocytes 81 273
## 1772066100_B01 pyramidal CA1 108 191
cellInfo <- data.frame(cellInfo)
cellTypeColumn <- "Class"
colnames(cellInfo)[which(colnames(cellInfo)==cellTypeColumn)] <- "CellType"
head(cellInfo)
## CellType nGene nUMI
## 1772066100_D04 interneurons 170 509
## 1772063062_G01 oligodendrocytes 152 443
## 1772060224_F07 microglia 218 737
## 1772071035_G09 pyramidal CA1 265 1068
## 1772067066_E12 oligodendrocytes 81 273
## 1772066100_B01 pyramidal CA1 108 191
#创建文件夹int
dir.create("int")
saveRDS(cellInfo, file="int/cellInfo.Rds")
#针对特定变量指定特定颜色
colVars <- list(CellType=c("microglia"="forestgreen",
"endothelial-mural"="darkorange",
"astrocytes_ependymal"="magenta4",
"oligodendrocytes"="hotpink",
"interneurons"="red3",
"pyramidal CA1"="skyblue",
"pyramidal SS"="darkblue"))
colVars$CellType <- colVars$CellType[intersect(names(colVars$CellType), cellInfo$CellType)]
saveRDS(colVars, file="int/colVars.Rds")
#看一下变量的颜色,这里是分成了5个细胞群,每个群用不同的颜色表示
plot.new(); legend(0,1, fill=colVars$CellType, legend=names(colVars$CellType))

2.3 初始化SCENIC设置
library(SCENIC)
org="mgi" # or hgnc, or dmel
dbDir="cisTarget_databases" # RcisTarget databases location
myDatasetTitle="SCENIC example on Mouse brain" # choose a name for your analysis
data(defaultDbNames)
dbs <- defaultDbNames[[org]]
scenicOptions <- initializeScenic(org=org, dbDir=dbDir, dbs=dbs, datasetTitle=myDatasetTitle, nCores=10)
## Motif databases selected:
## mm9-500bp-upstream-7species.mc9nr.feather
## mm9-tss-centered-10kb-7species.mc9nr.feather
scenicOptions@inputDatasetInfo$cellInfo <- "int/cellInfo.Rds"
scenicOptions@inputDatasetInfo$colVars <- "int/colVars.Rds"
saveRDS(scenicOptions, file="int/scenicOptions.Rds")
3. 构建基因共表达网络
这一步的目的是基于表达数据推断潜在的转录因子靶点。输入文件是过滤后的表达矩阵,一系列转录因子;输出是相关性矩阵,用于后续构建共表达模型(runSCENIC_1_coexNetwork2modules())。
这里有两种方法供选择:GENIE3(在R当中进行操作,能够鉴定非线性关系,耗时,计算量很大)和GRNboost(和前面一种方法类似,但耗时少,适合大数据)
3.1 过滤表达矩阵
#Filter by the total number of reads per gene
#Filter by the number of cells in which the gene is detected
#only the genes that are available in RcisTarget databases will be kept
genesKept <- geneFiltering(exprMat, scenicOptions=scenicOptions,
minCountsPerGene=3*.01*ncol(exprMat),
minSamples=ncol(exprMat)*.01)
exprMat_filtered <- exprMat[genesKept, ]
## [1] 770 200
#删除不需要的
rm(exprMat)
3.2 相关性
GENIE3/GRNboost能够检测到正相关和负相关的基因,为了将这两种相关性区分开,进行这一步处理。
#This step can be run either before/after or simultaneously to GENIE3/GRNBoost
runCorrelation(exprMat_filtered, scenicOptions)
3.3 GENIE3
To run GRNBoost (in Python) instead of GENIE3. See ?exportsForGRNBoost for details
这一步需要时间很长,可以另外开一个窗口单独运行这一步
# Optional: add log (if it is not logged/normalized already)
exprMat_filtered <- log2(exprMat_filtered+1)
# Run GENIE3
runGenie3(exprMat_filtered, scenicOptions)
4 构建并计算基因调控网络
library(SCENIC)
scenicOptions <- readRDS("int/scenicOptions.Rds")
scenicOptions@settings$verbose <- TRUE
scenicOptions@settings$nCores <- 10
scenicOptions@settings$seed <- 123
#这里为了计算方便,就选择了一个库
scenicOptions@settings$dbs <- scenicOptions@settings$dbs["10kb"] # For toy run
runSCENIC_1_coexNetwork2modules(scenicOptions)
runSCENIC_2_createRegulons(scenicOptions, coexMethod=c("top5perTarget")) #** Only for toy run!!
runSCENIC_3_scoreCells(scenicOptions, exprMat_filtered)
5 可选步骤:
5.1 将network activity转换成ON/OFF格式
#这一步是将前面得到的结果(AUC threshold)在shiny中进行可视化,你可以在其中手动调整阈值
aucellApp <- plotTsne_AUCellApp(scenicOptions, logMat)
savedSelections <- shiny::runApp(aucellApp)
# 感觉就是要更好的将细胞区分开,Save the modified thresholds:
newThresholds <- savedSelections$thresholds
scenicOptions@fileNames$int["aucell_thresholds",1] <- "int/newThresholds.Rds"
saveRDS(newThresholds, file=getIntName(scenicOptions, "aucell_thresholds"))
saveRDS(scenicOptions, file="int/scenicOptions.Rds")
#调整好新的阈值后,运行
runSCENIC_4_aucell_binarize(scenicOptions)
5.2 根据regulon activity 聚类降维
#这一步关键的参数“number of PCs” and “perplexity” (expected running time: few minutes to hours, depending on the number of cells)
nPcs <- c(5)
scenicOptions@settings$seed <- 123 # same seed for all of them
# Run t-SNE with different settings:
fileNames <- tsneAUC(scenicOptions, aucType="AUC", nPcs=nPcs, perpl=c(5,15,50))
fileNames <- tsneAUC(scenicOptions, aucType="AUC", nPcs=nPcs, perpl=c(5,15,50), onlyHighConf=TRUE, filePrefix="int/tSNE_oHC")
# Plot as pdf (individual files in int/):
fileNames <- paste0("int/",grep(".Rds", grep("tSNE_", list.files("int"), value=T), value=T))
# Using only "high-confidence" regulons (normally similar)
par(mfrow=c(3,3))
fileNames <- paste0("int/",grep(".Rds", grep("tSNE_oHC_AUC", list.files("int"), value=T, perl = T), value=T))
plotTsne_compareSettings(fileNames, scenicOptions, showLegend=FALSE, varName="CellType", cex=.5)
#下面会出图,根据细胞聚类结果选择好的参数值
scenicOptions@settings$defaultTsne$aucType <- "AUC"
scenicOptions@settings$defaultTsne$dims <- 5
scenicOptions@settings$defaultTsne$perpl <- 15
saveRDS(scenicOptions, file="int/scenicOptions.Rds")

6 对产生的中间结果进行进一步探索
6.1 Projection the AUC and TF expression onto t-SNEs
# 首先产生AUCell的交互式界面
#?可能也是对阈值进行调整
logMat <- exprMat # Better if it is logged/normalized
aucellApp <- plotTsne_AUCellApp(scenicOptions, logMat) # default t-SNE
savedSelections <- shiny::runApp(aucellApp)
#这里找到保存的rds对象的路径
print(tsneFileName(scenicOptions))
## [1] "int/tSNE_AUC_05pcs_15perpl.Rds"
tSNE_scenic <- readRDS(tsneFileName(scenicOptions))
aucell_regulonAUC <- loadInt(scenicOptions, "aucell_regulonAUC")
# Show TF expression:
par(mfrow=c(2,3))
AUCell::AUCell_plotTSNE(tSNE_scenic$Y, exprMat, aucell_regulonAUC[onlyNonDuplicatedExtended(rownames(aucell_regulonAUC))[c("Dlx5", "Sox10", "Sox9","Irf1", "Stat6")],], plots="Expression")

6.2 Density plot to detect most likely stable states (higher-density areas in the t-SNE)
library(KernSmooth)
library(RColorBrewer)
dens2d <- bkde2D(tSNE_scenic$Y, 1)$fhat
image(dens2d, col=brewer.pal(9, "YlOrBr"), axes=FALSE)
contour(dens2d, add=TRUE, nlevels=5, drawlabels=FALSE)

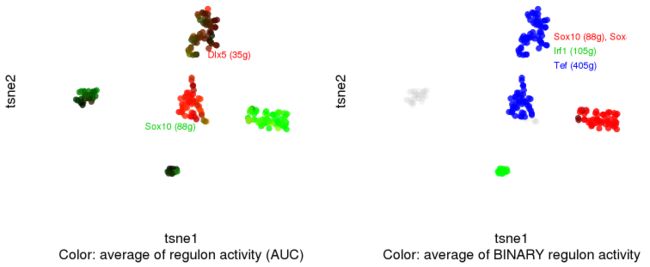
6.3 Show several regulons simultaneously
par(mfrow=c(1,2))
regulonNames <- c( "Dlx5","Sox10")
cellCol <- plotTsne_rgb(scenicOptions, regulonNames, aucType="AUC", aucMaxContrast=0.6)
text(0, 10, attr(cellCol,"red"), col="red", cex=.7, pos=4)
text(-20,-10, attr(cellCol,"green"), col="green3", cex=.7, pos=4)
regulonNames <- list(red=c("Sox10", "Sox8"),
green=c("Irf1"),
blue=c( "Tef"))
cellCol <- plotTsne_rgb(scenicOptions, regulonNames, aucType="Binary")
text(5, 15, attr(cellCol,"red"), col="red", cex=.7, pos=4)
text(5, 15-4, attr(cellCol,"green"), col="green3", cex=.7, pos=4)
text(5, 15-8, attr(cellCol,"blue"), col="blue", cex=.7, pos=4)
6.4 GRN: Regulon targets and motifs
#查看包含在regulon中的基因
regulons <- loadInt(scenicOptions, "regulons")
regulons[c("Dlx5", "Irf1")]
## $Dlx5
## [1] "2610203C20Rik" "Adamts17" "AI854703" "Arhgef10l" "Bahcc1" "Cirbp" "Cplx1" "Dlx1" "Gad1" "Gadd45gip1" "Hexim2" "Igf1"
## [13] "Iglon5" "Ltbp3" "Myt1" "Npas1" "Nxph1" "Peli2" "Plekha6" "Prkab1" "Ptchd2" "Racgap1" "Rgs8" "Robo1"
## [25] "Rpl34" "Sema3c" "Shisa9" "Slc12a5" "Slc39a6" "Spns2" "Stox2" "Syt6" "Unc5d" "Wnt5a"
##
## $Irf1
## [1] "4930523C07Rik" "Acsl5" "Adrb1" "Ahdc1" "Ahnak" "Akap1" "Anxa2" "Arhgap31" "Arhgef10l" "Arhgef19" "Atg14" "B2m"
## [13] "Bank1" "Bcl2a1b" "Cald1" "Capsl" "Ccl2" "Ccl3" "Ccl7" "Ccnd1" "Ccnd3" "Cd163" "Cd48" "Cited2"
## [25] "Cmtm6" "Ctgf" "Ctnnd1" "Ctss" "Ddr2" "E130114P18Rik" "Egfr" "Ehd1" "Esam" "Fcer1g" "Fcgr1" "Fgf14"
## [37] "Fstl4" "Fyb" "Gabrb1" "Gadd45g" "Gja1" "Gpr34" "Gramd3" "H2-K1" "Hfe" "Il6ra" "Irf1" "Itgb5"
## [49] "Kcne4" "Kcnj16" "Kcnj2" "Laptm5" "Lhfp" "Map3k5" "Mdk" "Med13" "Midn" "Mr1" "Ms4a7" "Msr1"
## [61] "Myh9" "Nin" "Nptx1" "Osbpl6" "P2ry13" "Parp12" "Peli2" "Plcl2" "Plekha6" "Prkab1" "Rab3il1" "Rgs5"
## [73] "Rhobtb2" "Rnase4" "Sepp1" "Sertad2" "Sgk3" "Slamf9" "Slc4a4" "Slc7a7" "Slco5a1" "Slitrk2" "Soat1" "Srgn"
## [85] "St3gal6" "St5" "St8sia4" "Stard8" "Stat6" "Stk17b" "Tapbp" "Tbxas1" "Tgfa" "Tgfb3" "Tmem100" "Tnfaip3"
## [97] "Tnfaip8" "Trib1" "Trpm7" "Txnip" "Usp2" "Vgll4" "Vps37b" "Zfp36l2" "Zfp703"
#注意,只有超过十个基因的regulons才会被AUCell计算
regulons <- loadInt(scenicOptions, "aucell_regulons")
head(cbind(onlyNonDuplicatedExtended(names(regulons))))
## [,1]
## Tef "Tef (405g)"
## Dlx5 "Dlx5 (35g)"
## Sox9 "Sox9 (150g)"
## Sox8 "Sox8 (97g)"
## Sox10 "Sox10 (88g)"
## Irf1 "Irf1 (105g)"
#查看TF与靶点之间的联系
regulonTargetsInfo <- loadInt(scenicOptions, "regulonTargetsInfo")
tableSubset <- regulonTargetsInfo[TF=="Stat6" & highConfAnnot==TRUE]
viewMotifs(tableSubset)
6.5 Regulators for clusters or known cell types
#Average Regulon Activity by cluster
#聚类这里的cluster参数选择可以是CellType,也可以是Seurat对象里面的特定分组
#这里除了可以选择aucell_regulonAUC,还可以选择aucell_binary_nonDupl
regulonAUC <- loadInt(scenicOptions, "aucell_regulonAUC")
regulonAUC <- regulonAUC[onlyNonDuplicatedExtended(rownames(regulonAUC)),]
regulonActivity_byCellType <- sapply(split(rownames(cellInfo), cellInfo$CellType),
function(cells) rowMeans(getAUC(regulonAUC)[,cells]))
regulonActivity_byCellType_Scaled <- t(scale(t(regulonActivity_byCellType), center = T, scale=T))
pheatmap::pheatmap(regulonActivity_byCellType_Scaled, #fontsize_row=3,
color=colorRampPalette(c("blue","white","red"))(100), breaks=seq(-3, 3, length.out = 100),
treeheight_row=10, treeheight_col=10, border_color=NA)
