NGS数据格式梳理02-SAM/BAM格式最详细解读
本篇是自己学习SAM和SAMtag的资料心得,为防自己理解有误,误导他人,参考资料(文中“[ ]”中的数字对应文末的参考文献)都有加上,不足之处欢迎指正。
目录
SAM/BAM格式简介[1]
术语与概念理解[3][4]
标头部分(header section)
比对信息部分(alignment section)
比对部分概述
比对部分详述[4]
第一列、QNAME
第二列、FLAG
第三列、RNAME
第四列、POS
第五列、MAPQ
第六列、CIGAR
第七列、RNEXT
第八列、PNEXT
第九列、TLEN
第十列、SEQ
第十一列、QUAL
第十二列之后,Optional fields
1.1 Additional Template and Mapping data(一些比对信息)
1.2 Metadata(这部分内容和 SAM中header section部分相关,描述read测序相关信息)
1.3 Barcodes(UMI/单细胞测序cell barcode)
1.4 Original data
1.5 Annotation and Padding
1.6 Technology-specifific data
参考资料
我的公众号
SAM/BAM格式简介[1]
- SAM存储格式发明的目的:使得不同平台下机数据,经过不同比对软件后有一个统一的存储格式。
- SAM(Sequence Alignment/Map format简写)格式文件,存储测序数据和参考基因组比对结果的文件,每行以table键分割,包含标头部分(header section)和比对部分(alignment section)见下图。
- BAM(Binary Alignment/Map format简写)格式文件,SAM的二进制格式文件,通过BGZF library参考库压缩而成。

图一,SAM/BAM格式简介[2]
术语与概念理解[3][4]
该部分有助于后文SAM格式理解,后文反复出现如下概念。
- 模板(Template):一段DNA/RNA序列,它的一部分在测序仪上被测序,或被从原始序列中组装。(意思就是:我们通过测序仪测序的那段序列,或者通过组装原始序列得到的更长的序列,就是模板的一部分)。(从后文来看,对于Illumina双端测序来说,template指的就是插入片段)
- 片段(Segment):一段连续的序列或子序列(subsequence)(从上下文来看,segment既可以指一条完整的read,也可以指read的一部分);
- 读段(Read):一段来自测序仪的原始序列。read可以包含多个片段(一条read在比对过程中可能会被拆分成几段,对应到参考序列不同的位置上。read被拆分后形成的片段即为segment)。对于测序数据,reads根据测序顺序进行编号;
- 线性比对(Linear alignment):一条比对到参考序列上的read可能会有插入、缺失、skips和切除(clipping),但只要没有方向的改变(例如,read的一部分比对到了正义链上,另一部分比对到了反义链上),就是Linear alignment。一个线性比对结果可以代表一个SAM记录;(意思似乎是:一条SAM记录能且只能保存一个线性比对结果)
- 嵌合比对(Chimeric alignment):不是线性比对的比对。嵌合比对中包含了一套没有大范围重叠的线性比对(嵌合比对中的每一个片段都是线性比对。关于大范围重叠的说法是为了和多重比对区分)。一般地,嵌合比对中的一个线性比对被认为是“有代表性的比对”(representative alignment),而其他的线性比对被称为补充的(supplementary),用补充比对标志(supplementary alignment flag)加以区别(representative和supplementary成一对,对应嵌合比对)。嵌合比对的所有SAM记录有相同的QNAME,其flag值的0x40和0x80位都相同(见1.4节)(0x40位和0x80位分别表示模板中的第一个片段和最后一个片段,为什么会都相同呢?总要有一个是第一个片段,总要有一个是最后一个片段吧,它俩的0x40位和0x80位不应该相同啊?)。哪个线性比对被视为有代表性是任意选择的。(可见嵌合比对中,各个segments的独立性更强:都不在双链的同一条链了。另外,如果一条read的不同部分比对到了不同的染色体上,那肯定也是嵌合比对了,因为不同染色体之间讨论方向相同是没有意义的,肯定不可能是线性比对了。)
- read比对(read alignment):能代表一条read的比对结果的线性比对或嵌合比对;
- 多重比对(Multiple mapping):由于重复序列等情况的存在,一条read在参考基因组上的正确位置可能无法确定。在这种情况下,一条read可能会有多种比对结果,其中一种被视为主要的(primary),所有其他的比对结果的SAM记录的flag标志中都会有一个“次要(secondary)比对结果”的标志。所有这些SAM记录拥有相同的QNAME,flag标志的0x40位和0x80位有相同的值。一般被指定为“主要”的比对结果是最佳比对,如果都是最佳比对,则任意指定一条(primary和secondary成一对,对应多重比对)。(原文注释:嵌合比对主要由结构变异、基因融合、组装错误、RNA测序或实验过程中的一些原因造成,更经常出现在长reads中(长read有利于检测嵌合比对。这就是为什么三代测序是检测染色体结构变异的更有力工具)。嵌合比对中的线性比对之间没有大片段的重叠,每个线性比对有较高的mapping质量值,可以用于SNP/INDEL的检测;而多重比对主要是序列重复造成的,不经常出现在长reads中。如果一条read有多重比对的情况,所有的比对互相之间几乎完全完全重叠。除了一个最佳比对外,所有其他比对的质量值都<3,且会被大多数SNP/INDEL检测软件忽略)。
- 以1为起始的坐标系(1-based coordinate system):序列的第一位是1的坐标系。在这种坐标系中,一个区域用闭区间表示。例如,第三位和第七位碱基之间的区域表示为[3,7]。SAM, VCF, GFF和Wiggle格式使用以1为起始的坐标系;
- 以0为起始的坐标系(0-based coordinate system):序列的第一位是0的坐标系。在这种坐标系中,一个区域用左闭右开区间表示。例如,第三位和第七位碱基之间的区域表示为[2,7)(原文如此。难道不应该是[3,8)么?不应该。以0为起始,第三位对应的索引号是2,第七位对应的索引号是6,所以索引号[2,7)对应了第三位-第七位碱基。当时脑子糊涂了,没搞清文中说的意思)。BAM, BCFv2, BED和PSL格式使用以0为起始的坐标系;
- Phred scale:如果一个概率值0
标头部分(header section)
该部分为SAM/BAM的注释部分,该部分并非必须,可以省略。每一行都以@符开头,后面跟着两个大写字母,每个字段之间以\t分割,每个字段遵循(TAG:Value)的格式(@CO开头的行除外)。每行可以使用以下正则表达式表示:/^@(HD|SQ|RG|PG)(\t[A-Za-z][A-Za-z0-9]:[ -~]+)+$/ or /^@CO\t.*/,@后紧跟的两个大写字母主要有HD,SQ,RG,PG和CO五类,前四类常用如下表,其中加了*号的表示该标签必须存在,例如@HD这个标签存在时,VN必须同时存在,详细介绍如下[3][4]。
| @HD |
该SAM文件的版本,SAM的sort方式 |
|
| VN* |
Version简写,SAM文件的版本。 |
|
| SO |
Sorting order of alignments 简写,比对时的sort顺序,包含如下几类:unknown(默认的), unsorted, queryname和coordinate。coordinate类先以SAM中第三列(RNAME)排序,即@SQ中的SN顺序;如果RNAME相同,则再以SAM中第四列(POS字段)排序。RNAME为*时,则该read放在其它结果之后,顺序任意。 |
|
| GO |
Grouping of alignments 简写,比对结果的分组信息,相似的比对结果聚集到一起,有以下几类分组方法:none(默认的),query(根据QNAME分组),reference(根据RNAME/POS分组)。 |
|
| SS |
Sub-sorting order of alignments 简写,比对时的sub-sort排序,即当排序方式不在上面SO中的四类时,以SS方式排序,例如 @HD SO:unsorted SS:unsorted:MI:coordinate。 |
|
| @SQ |
比对使用参考基因组信息。@SQ的顺序即为参考基因组中染色体的sort顺序。例如,@SQ SN:chr22 LN:51304566 |
|
| SN* |
Reference sequence name 简写,参考序列名称。每行SN标签独一无二,RNAME和RNEXT会使用到。 |
|
| LN* |
Reference sequence length 简写,参考序列长度,范围[1,2^31-1]。 |
|
| AH |
Alternate locus 简写,表明参考基因组版本是基于alt_scaffold时,还有一种参考基因组是基于primary_assembly的。 |
|
| AN |
Alternative reference sequence names 简写。 |
|
| AS |
Genome assembly identififier,参考基因组组装ID。 |
|
| DS |
Description. UTF-8 encoding may be used. |
|
| M5 |
参考基因组MD5校验,allows reference sequences to be uniquely identifified through the MD5 digest of the sequence itself. |
|
| SP |
物种 |
|
| MT |
分子拓扑信息(molecule topology),默认是线性,还有环状。 |
|
| UR |
URI of the sequence,参考基因组的路径,‘http:’ or ‘ftp:’开头。 |
|
| @RG |
Read group 简写,该标签重点关注sample sam 中reads测序信息。 |
|
| ID* |
每个sample SAM对应一个独一无二的ID。 |
|
| CN |
产生sample read的测序中心的名字。 |
|
| DS |
描述(Description) |
|
| DT |
sample read测序日期 |
|
| FO |
流程顺序 flow order。 |
|
| KS |
The array of nucleotide bases that correspond to the key sequence of each read. |
|
| LB |
文库名称 |
|
| PG |
Programs used for processing the read group. |
|
| PI |
Predicted median insert size. |
|
| PL |
测序平台,例如ILLUMINA |
|
| PM |
平台模式,提供平台/技术进一步细节的自由格式的文本。 |
|
| PU |
测序平台详细信息(例如flowcell号,lane号(对Illumina)。 |
|
| SM |
样本名称。如果混样的话,就用pool名称。 |
|
| @PG |
该标签关注使用何种比对软件及生成当前sam文件的命令行。 |
|
| ID* |
比对软件的唯一ID。 |
|
| PN |
软件的名称,例如bwa。 |
|
| CL |
比对命令行,例如bwa samse hg19.fasta sample.fastq -f sample.sam |
|
| PP |
前一个程序的标识。 |
|
| DS |
描述 |
|
| VN |
比对软件版本号。 |
|
| @CO |
其它注释信息。 |
|
比对信息部分(alignment section)
-
比对部分概述
该部分是SAM文件的核心部分,每一行代表一个序列的线性比对(linear alignment of a segment),每行包含前11个必需字段,和第12个字段后多个可选字段,使用TAB-separated分割,当某个字段信息缺省时,如果字段是字符串型以*替代,如果字段是整型以‘0’来替代,下表为11个必需字段含义的概述[4]:
| 字段所在列号 |
区段 |
类型 |
正则表达式 |
简要概述 |
| 1 |
QNAME |
字符串 |
[!-?A-~]{1,254} |
需要比对的序列名称(FASTQ中第一行) |
| 2 |
FLAG |
整型 |
[0,2^16-1] |
FLAG的位操作符 |
| 3 |
RNAME |
字符串 |
\*|[!-()+-<>-~][!-~]* |
参考基序列的名称 |
| 4 |
POS |
整型 |
[0,2^31-1] |
序列比对到参考序列中的起始位置坐标(以1为起始) |
| 5 |
MAPQ |
整型 |
[0,2^8-1] |
比对质量值 |
| 6 |
CIGAR |
字符串 |
\*|([0-9]+[MIDNSHPX=])+ |
CIGAR字符串 |
| 7 |
RNEXT |
字符串 |
\*|=|[!-()+-<>-~][!-~]* |
双端测序中另外一个read比对的参考序列名称 |
| 8 |
PNEXT |
整型 |
[0,2^31-1] |
双端测序中另外一个read比对到参考序列中的起始位置坐标 |
| 9 |
TLEN |
整型 |
[-2^31+1,2^31-1] |
建库时打断的长度 |
| 10 |
SEQ |
字符串 |
\*|[A-Za-z=.]+ |
序列碱基信息(FASTQ中第三行) |
| 11 |
QUAL |
字符串 |
[!-~]+ |
SEQ字段对应的ASCII码质量字符(FASTQ中第四行) |
-
比对部分详述[4]
-
第一列、QNAME
被比对序列的名称(query template name),如果QNAME唯一,则序列被认为来源于同一模板;‘*’表示该字段缺省;一般情况下,该字段为FASTQ文件的第一行信息;嵌合(Chimeric alignment)比对或者多次比对(Multiple mapping)的序列会导致一个QNAME在SAM中多次出现。
-
第二列、FLAG
SAM中显示的是下图中第一列值或者第一列中的数值和,当显示的是下表中第一列数值时,意义为Description所列出,如果是多个数值和,意义为Description多行意义汇总,常用的意义见下表:

- Description意义
1 :该read使用双端测序,单端测序为0;
2: 该read和完全比对到参考序列;
4: 该read没有比对到参考序列;
8: 双端序列的另外一条序列没有比对上参考序列(read1或者read2);
16:该read比对到参考序列的负链上(该read反向互补比对到参考序列);
32 :该read的另一条read比对到参考序列的负链上;
64 :双端测序 read1;
128 : 双端测序read2;
256: 该read不是最佳的比对序列,一条read能比对到参考序列的多个位置,只有一个是最佳的比对位置,其他都是次要的;
512: 该read在过滤(碱基质量,测序平台等指标)时没通过;
1024: PCR(文库构建时)或者仪器(测序时)导致的重复序列;
2048: 该read可能存在嵌合(发生在PCR过程中),当前比对部分只是read的一部分;
- 如果FLAG不在上表第一列,可以使用如下两个网站查询:
网站1:http://https://broadinstitute.github.io/picard/explain-flags.html
例如,FLAG 88=8(0x8对应值)+16(0x10对应值)+64(0x40对应值),该FLAG值意义为三个意义的汇总。

网站2:https://www.samformat.info/sam-format-flag
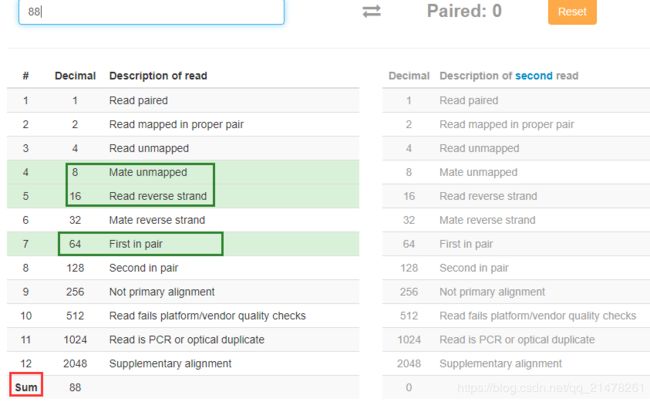
- 另外一些常用FLAG
- One of the reads is unmapped(双端reads只有一条reads比对上):
73, 133, 89, 121, 165, 181, 101, 117, 153, 185, 69, 137
- Both reads are unmapped(双端reads都没比对上):
77, 141
- Mapped within the insert size and in correct orientation(reads比对上了,大小方向均对):
99, 147, 83, 163
- Mapped within the insert size but in wrong orientation(比对上了,但是方向不对):
67, 131, 115, 179
- Mapped uniquely, but with wrong insert size(唯一比对,但是大小不对):
81, 161, 97, 145, 65, 129, 113, 177
-
第三列、RNAME
Reference sequence NAME of the alignment,比对时参考序列的名称,一般是染色体号(如果物种为人,则为chr1~chr22,chrX,chrY,chrM)。RNAME(如果不是‘*’)必须在header section部分@SQ中SN标签后出现。如果没有比对上参考基因组,用‘*’来表示。如果RNAME值是‘*’,则后面POS和CIGAR也将没有值。
-
第四列、POS
该read比对到参考基因组的位置坐标,最小为1(1-based leftmost)。该read如果没有比对上参考序列,则RNAME和CIGAR也无值。
-
第五列、MAPQ
对应参考序列的质量(MAPing Quality),比对的质量分数,越高说明该read比对到参考基因组上的位置越准确。其值等于-10 lg Probility (错配概率),得出值后四舍五入的整数就是MAPQ值。如果该值是255,则说明对应质量无效。例如,MAPQ为20,即Q20,错误率为0.01,20 = -10log10(0.01) = -10*(-2)。
-
第六列、CIGAR
Compact Idiosyncratic Gapped Alignment Representation的简写,描述read与参考序列的比对具体情况信息。CIGAR中的数字代表碱基的个数,字符的含义见下表:
| 字符 |
BAM |
描述 |
序列是否出现在read中 |
序列是否出现在参考序列中 |
| M |
0 |
比对匹配(match) |
yes |
yes |
| I |
1 |
插入(insertion) |
yes |
no |
| D |
2 |
缺失(deletion) |
no |
yes |
| N |
3 |
跳过参考序列,转录组中表示遇到内含子 |
no |
yes |
| S |
4 |
软跳过(soft clipping),比对时跳过read中部分序列,不改变read长度 |
yes |
no |
| H |
5 |
硬跳过(hard clipping),比对时直接剪掉部分序列,read长度被改变,发生在比对开始或者结束时 |
no |
no |
| P |
6 |
padding,read比对时中间跳过参考序列部分区域 |
no |
no |
| = |
7 |
该read完全匹配 |
yes |
yes |
| X |
8 |
该read不匹配 |
yes |
yes |
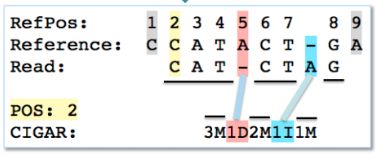
举例:3M1D2M1I1M:3个碱基匹配(M)(3M)、接下来1个碱基缺失(D)、接下来2个匹配(2M)、接下来1个碱基插入(1I)、接下来1个碱基匹配(1M),如下图:
-
第七列、RNEXT
双端测序中另外一条read比对的参考序列的名称,单端测序此处为0,RNEXT(如果不是*或者=,*是完全没有比对上,=是完全比对)必须在header section部分@SQ中SN标签后出现。第3和第7列,可以用来判断某条read是否比对成功到了参考序列上,read1和read2是否比对到同一条参考染色体上。
-
第八列、PNEXT
双端测序中,是指另外一条read比对到参考基因组的位置坐标,最小为1(1-based leftmost)。
-
第九列、TLEN
文库长度,insert DNA size。

-
第十列、SEQ
read 碱基序列,FASTQ的第二行。
-
第十一列、QUAL
FASTQ的第四行。
-
第十二列之后,Optional fields
可选的自定义区域(Optional fields),可能有多列,多列间使用\t隔开,并不是每行都存在这些列。
XT:A:R NM:i:0 X0:i:4 XM:i:0 XO:i:0 XG:i:0 MD:Z:50 XA:Z:chr1,+102573964,50M,0
XT:A:U NM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
XT:A:U NM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
#该行该列没有内容
XT:A:U NM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:50
每列格式为TAG:TYPE:VALUE,其中
- TAG为两个大写字母;
- TYPE可以由如下格式A (character), B (general array), f (real number), H (hexadecimal array), i (integer), or Z (string);
- VALUE ,内容与TYPE相关,TYPE为i时VALUE为整数,以此类推;
TAG详细介绍
可分为6类,详细介绍如下[6]:
-
1.1 Additional Template and Mapping data(一些比对信息)
AM:i:score The smallest template-independent mapping quality of any segment in the same template as
this read. (See also SM.)
AS:i:score Alignment score generated by aligner.
BQ:Z:qualities Offffset to base alignment quality (BAQ), of the same length as the read sequence. At the
i-th read base, BAQi = Qi
(BQi
64) where Qi is the i-th base quality.
CC:Z:rname Reference name of the next hit; ‘=’ for the same chromosome.
CG:B:I,encodedCigar Real CIGAR in its binary form if (and only if) it contains >65535 operations. This
is a BAM fifile only tag as a workaround of BAM’s incapability to store long CIGARs in the standard
way. SAM and CRAM fifiles created with updated tools aware of the workaround are not expected to
contain this tag. See also the footnote in Section 4.2 of the SAM spec for details.
2CP:i:pos Leftmost coordinate of the next hit.
E2:Z:bases The 2nd most likely base calls. Same encoding and same length as SEQ. See also U2 for
associated quality values.
FI:i:int The index of segment in the template.
FS:Z:str Segment suffiffiffix.
H0:i:count Number of perfect hits.
H1:i:count Number of 1-difffference hits (see also NM).
H2:i:count Number of 2-difffference hits.
HI:i:i Query hit index, indicating the alignment record is the i-th one stored in SAM.
IH:i:count Number of alignments stored in the fifile that contain the query in the current record.
MC:Z:cigar CIGAR string for mate/next segment.
MD:Z:[0-9]+(([A-Z]|\^[A-Z]+)[0-9]+)* String for mismatching positions.
The MD fifield aims to achieve SNP/indel calling without looking at the reference. For example, a string
‘10A5^AC6’ means from the leftmost reference base in the alignment, there are 10 matches followed
by an A on the reference which is difffferent from the aligned read base; the next 5 reference bases are
matches followed by a 2bp deletion from the reference; the deleted sequence is AC; the last 6 bases are
matches. The MD fifield ought to match the CIGAR string.
MQ:i:score Mapping quality of the mate/next segment.
NH:i:count Number of reported alignments that contain the query in the current record.
NM:i:count Number of difffferences (mismatches plus inserted and deleted bases) between the sequence and reference, counting only (case-insensitive) A, C, G and T bases in sequence and reference as potential matches, with everything else being a mismatch(可以结合CIGAR字段计算错配碱基个数). Note this means that ambiguity codes in both
sequence and reference that match each other, such as ‘N’ in both, or compatible codes such as ‘A’ and
‘R’, are still counted as mismatches. The special sequence base ‘=’ will always be considered to be a
match, even if the reference is ambiguous at that point. Alignment reference skips, padding, soft and
hard clipping (‘N’, ‘P’, ‘S’ and ‘H’ CIGAR operations) do not count as mismatches, but insertions and
deletions count as one mismatch per base.Note that historically this has been ill-defifined and both data and tools exist that disagree with this defifinition.
PQ:i:score Phred likelihood of the template, conditional on the mapping locations of both/all segments
being correct.
Q2:Z:qualities Phred quality of the mate/next segment sequence in the R2 tag. Same encoding as QUAL.
R2:Z:bases Sequence of the mate/next segment in the template. See also Q2 for any associated quality
values.
SA:Z:(rname ,pos ,strand ,CIGAR ,mapQ ,NM ;)+ Other canonical alignments in a chimeric alignment, for
matted as a semicolon-delimited list. Each element in the list represents a part of the chimeric align
ment. Conventionally, at a supplementary line, the fifirst element points to the primary line. Strand is
either ‘+’ or ‘-’, indicating forward/reverse strand, corresponding to FLAG bit 0x10. Pos is a 1-based
coordinate.
SM:i:score Template-independent mapping quality, i.e., the mapping quality if the read were mapped as
a single read rather than as part of a read pair or template.
3TC:i: The number of segments in the template.
TS:A:strand Strand (‘+’ or ‘-’) of the transcript to which the read has been mapped.
U2:Z: Phred probability of the 2nd call being wrong conditional on the best being wrong. The same
encoding and length as QUAL. See also E2 for associated base calls.
UQ:i: Phred likelihood of the segment, conditional on the mapping being correct.
-
1.2 Metadata(这部分内容和 SAM中header section部分相关,描述read测序相关信息)
RG:Z:readgroup The read group to which the read belongs. If @RG headers are present, then readgroup
must match the RG-ID fifield of one of the headers.
LB:Z:library The library from which the read has been sequenced. If @RG headers are present, then library
must match the RG-LB fifield of one of the headers.
PG:Z:program id Program. Value matches the header PG-ID tag if @PG is present.
PU:Z:platformunit The platform unit in which the read was sequenced. If @RG headers are present, then
platformunit must match the RG-PU fifield of one of the headers.
CO:Z:text Free-text comments.
-
1.3 Barcodes(UMI/单细胞测序cell barcode)
DNA barcodes can be used to identify the provenance of the underlying reads. There are currently three
varieties of barcodes that may co-exist: Sample Barcode, Cell Barcode, and Unique Molecular Identififier
(UMI).
• Despite its name, the Sample Barcode identififies the Library and allows multiple libraries to be combined
and sequenced together. After sequencing, the reads can be separated according to this barcode and
placed in difffferent “read groups” each corresponding to a library. Since the library was generated from
a sample, knowing the library should inform of the sample. The barcode itself can be included in the
PU fifield in the RG header line. Since the PU fifield should be globally unique, it is advisable to include
specifific information such as flflowcell barcode and lane. It is not recommended to use the barcode as
the ID fifield of the RG header line, as some tools modify this fifield (e.g., when merging fifiles).
• The Cell Barcode is similar to the sample barcode but there is (normally) no control over the assignment
of cells to barcodes (whose sequence could be random or predetermined). The Cell Barcode can help
identify when reads come from difffferent cells in a “single-cell” sequencing experiment.(在单细胞测序中,追溯read来源的标签)
• The UMI is intended to identify the (single- or double-stranded) molecule at the time that the barcode
was introduced. This can be used to inform duplicate marking and make consensus calling in ultra
deep sequencing. Additionally, the UMI can be used to (informatically) link reads that were generated
from the same long molecule, enabling long-range phasing and better informed mapping. In some
experimental setups opposite strands of the same double-stranded DNA molecule get related barcodes.
These templates can also be considered duplicates even though technically they may have difffferent
UMIs. Multiple UMIs can be added by a protocol, possibly at difffferent time-points, which means that
specifific knowledge of the protocol may be needed in order to analyze the resulting data correctly.(UMI信标签,RNA-seq中UMI可以对原始的 RNA 分子进行“绝对定量”)
BC:Z:sequence Barcode sequence (Identifying the sample/library), with any quality scores (optionally)
stored in the QT tag. The BC tag should match the QT tag in length. In the case of multiple unique
molecular identififiers (e.g., one on each end of the template) the recommended implementation con
catenates all the barcodes and places a hyphen (‘-’) between the barcodes from the same template.
QT:Z:qualities Phred quality of the sample barcode sequence in the BC tag. Same encoding as QUAL,
i.e., Phred score + 33. In the case of multiple unique molecular identififiers (e.g., one on each end of
the template) the recommended implementation concatenates all the quality strings with spaces (‘ ’)
between the difffferent strings from the same template.
4CB:Z:str Cell identififier, consisting of the optionally-corrected cellular barcode sequence and an optional
suffiffiffix. The sequence part is similar to the CR tag, but may have had sequencing errors etc corrected.
This may be followed by a suffiffiffix consisting of a hyphen (‘-’) and one or more alphanumeric characters to form an identififier. In the case of the cellular barcode (CR) being based on multiple barcode sequences
the recommended implementation concatenates all the (corrected or uncorrected) barcodes with a
hyphen (‘-’) between the difffferent barcodes. Sequencing errors etc aside, all reads from a single cell
are expected to have the same CB tag.
CR:Z:sequence+ Cellular barcode. The uncorrected sequence bases of the cellular barcode as reported
by the sequencing machine, with the corresponding base quality scores (optionally) stored in CY. Se
quencing errors etc aside, all reads with the same CR tag likely derive from the same cell. In the case
of the cellular barcode being based on multiple barcode sequences the recommended implementation
concatenates all the barcodes with a hyphen (‘-’) between the difffferent barcodes.
CY:Z:qualities+ Phred quality of the cellular barcode sequence in the CR tag. Same encoding as QUAL,
i.e., Phred score + 33. The lengths of the CY and CR tags must match. In the case of the cellular
barcode being based on multiple barcode sequences the recommended implementation concatenates all
the quality strings with with spaces (‘ ’) between the difffferent strings.
MI:Z:str Molecular Identififier. A unique ID within the SAM fifile for the source molecule from which this
read is derived. All reads with the same MI tag represent the group of reads derived from the same
source molecule.
OX:Z:sequence+ Raw (uncorrected) unique molecular identififier bases, with any quality scores (optionally)
stored in the BZ tag. In the case of multiple unique molecular identififiers (e.g., one on each end of the
template) the recommended implementation concatenates all the barcodes with a hyphen (‘-’) between
the difffferent barcodes.
BZ:Z:qualities+ Phred quality of the (uncorrected) unique molecular identififier sequence in the OX tag.
Same encoding as QUAL, i.e., Phred score + 33. The OX tags should match the BZ tag in length. In the
case of multiple unique molecular identififiers (e.g., one on each end of the template) the recommended
implementation concatenates all the quality strings with a space (‘ ’) between the difffferent strings.
RX:Z:sequence+ Sequence bases from the unique molecular identififier. These could be either corrected or
uncorrected. Unlike MI, the value may be non-unique in the fifile. Should be comprised of a sequence of
bases. In the case of multiple unique molecular identififiers (e.g., one on each end of the template) the
recommended implementation concatenates all the barcodes with a hyphen (‘-’) between the difffferent
barcodes.If the bases represent corrected bases, the original sequence can be stored in OX (similar to OQ storing the original qualities of bases.)
QX:Z:qualities+ Phred quality of the unique molecular identififier sequence in the RX tag. Same encoding
as QUAL, i.e., Phred score + 33. The qualities here may have been corrected (Raw bases and qualities
can be stored in OX and BZ respectively.) The lengths of the QX and the RX tags must match. In the
case of multiple unique molecular identififiers (e.g., one on each end of the template) the recommended
implementation concatenates all the quality strings with a space (‘ ’) between the difffferent strings.
-
1.4 Original data
OA:Z:(RNAME,POS,strand,CIGAR,MAPQ,NM ;)+ The original alignment information of the record
prior to realignment or unalignment by a subsequent tool. Each original alignment entry contains
the following six fifield values from the original record, generally in their textual SAM representations,
separated by commas (‘,’) and terminated by a semicolon (‘;’): RNAME, which must be explicit
(unlike RNEXT, ‘=’ may not be used here); 1-based POS; ‘+’ or ‘-’, indicating forward/reverse strand
respectively (as per bit 0x10 of FLAG); CIGAR; MAPQ; NM tag value, which may be omitted (though
the preceding comma must be retained).
5In the presence of an existing OA tag, a subsequent tool may append another original alignment entry
after the semicolon, adding to—rather than replacing—the existing OA information.
The OA fifield is designed to provide record-level information that can be useful for understanding the
provenance of the information in a record. It is not designed to provide a complete history of the
template alignment information. In particular, realignments resulting in the the removal of Secondary
or Supplementary records will cause the loss of all tags associated with those records, and may also
leave the SA tag in an invalid state.
OC:Z:cigar Original CIGAR, usually before realignment. Deprecated in favour of the more general OA.
OP:i:pos Original 1-based POS, usually before realignment. Deprecated in favour of the more general OA.
OQ:Z:qualities Original base quality, usually before recalibration. Same encoding as QUAL.
-
1.5 Annotation and Padding
The SAM format can be used to represent de novo assemblies , generally by using padded reference sequences and the annotation tags described here. See the Guide for Describing Assembly Sequences in the SAM Format Specifification for full details of this representation.
CT:Z:strand;type(;key(=value)?)*
Complete read annotation tag, used for consensus annotation dummy features.
The CT tag is intended primarily for annotation dummy reads, and consists of a strand, type and zero or
more key=value pairs, each separated with semicolons. The strand fifield has four values as in GFF3,2
and supplements FLAG bit 0x10 to allow unstranded (‘.’), and stranded but unknown strand (‘?’)
annotation. For these and annotation on the forward strand (strand set to ‘+’), do not set FLAG bit
0x10. For annotation on the reverse strand, set the strand to ‘-’ and set FLAG bit 0x10.
The type and any keys and their optional values are all percent encoded according to RFC3986 to
escape meta-characters ‘=’, ‘%’, ‘;’, ‘|’ or non-printable characters not matched by the isprint() macro
(with the C locale). For example a percent sign becomes ‘%25’.
PT:Z:annotag(\|annotag)*
where each annotag matches start;end;strand;type(;key(=value)?)* Read annotations for parts of the padded read sequence.The PT tag value has the format of a series of annotation tags separated by ‘|’, each annotating a sub-region of the read. Each tag consists of start, end, strand, type and zero or more key=value pairs,each separated with semicolons. Start and end are 1-based positions between one and the sum of the M/I/D/P/S/=/X CIGAR operators, i.e., SEQ length plus any pads. Note any editing of the CIGAR
string may require updating the PT tag coordinates, or even invalidate them. As in GFF3, strand is
one of ‘+’ for forward strand tags, ‘-’ for reverse strand, ‘.’ for unstranded or ‘?’ for stranded but unknown strand. The type and any keys and their optional values are all percent encoded as in the CT tag.
-
1.6 Technology-specifific data
FZ:B:S,intensities Flow signal intensities(测序拍照的光强度数据) on the original strand of the read, stored as (uint16 t)
round(value * 100.0).
1.6.1 Color space
CM:i:distance Edit distance between the color sequence and the color reference (see also NM).
CS:Z:sequence Color read sequence on the original strand of the read. The primer base must be included.
CQ:Z:qualities Color read quality on the original strand of the read. Same encoding as QUAL; same
length as CS.
-
2 Locally-defifined tags
You can freely add new tags. Note that tags starting with ‘X’, ‘Y’, or ‘Z’ and tags containing lowercase letters in either position are reserved for local use and will not be formally defifined in any future version of this specifification. If a new tag may be of general interest, it may be useful to have it added to this specifification. Additions can be proposed by opening a new issue at https://github.com/samtools/hts-specs/issues and/or by sending email to [email protected].
参考资料
[1] Li H, Handsaker B, Wysoker A, et al. The Sequence Alignment/Map format and SAMtools[J]. Bioinformatics, 2009, 25(16): 2078-2079.
[2] https://www.samformat.info/sam-format-flag
[3] http://note.youdao.com/share/?id=312fa04209cb87f7674de9a9544f329a&type=note#/
[4] https://samtools.github.io/hts-specs/SAMv1.pdf
[5] https://yulijia.net/slides/bioinfomatcis_for_medical_students/2019-07-31-A_beginners_guide_to_Call_SNPs_and_indels_Part_II.html#1
[6] http://samtools.github.io/hts-specs/SAMtags.pdf
我的公众号
力求详细专业介绍生信相关技术,欢迎一起学习!
