RNA-seq流程学习笔记(9)-使用RSeQC软件对生成的BAM文件进行质控
参考文章:
用RSeQC对比对后的转录组数据进行质控
高通量测序质控及可视化工具包RSeQC
RSeQC使用笔记
1. 质控的原因及相关软件
在A survey of best practices for RNA-seq data analysis里面,提到了人类基因组应该有70%~90%的比对率,并且多比对read(multi-mapping reads)数量要少。另外比对在外显子和所比对链(uniformity of read coverage on exons and the mapped strand)的覆盖度要保持一致。因此,可以对之前得到的BAM比对文件进行质检。
对BAM文件进行QC的软件包括:
Qualimap:对二代数据进行质控的综合软件
Picard:综合质控学习软件。
RSeQC是发表于2012年的一个RNA-Seq质控工具,属于python包。提供了一系列有用的小工具能够评估高通量测序尤其是RNA-seq数据。比如一些基本模块:检查序列质量、核酸组分偏性、PCR偏性、GC含量偏性,还有RNA-seq特异性模块:评估测序饱和度、映射读数分布、覆盖均匀性、链特异性、转录水平RNA完整性等。
2. RSeQC软件安装
参照文章:RNA-seq流程学习笔记(3)
查看Conda官网Index的RSeQC软件介绍,发现支持python3.6版本,因此直接使用Miniconda3安装, 安装完成后并没有RSeQC这个软件,而是增加了一些python命令,如下:
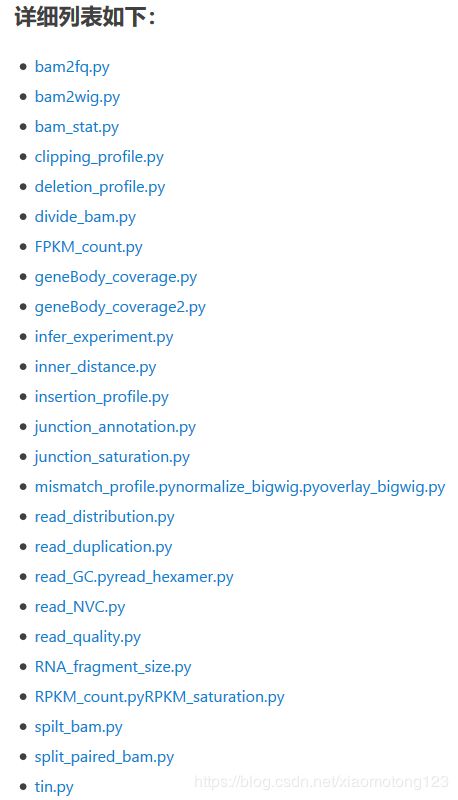
虽然该软件的使用命令非常多,但很多功能并不是用来诊断转录组测序的,所以不在我们的考虑范围内。
3.RSeQC处理4种文件格式:
- BED 格式:Tab 分割,12列的表示基因模型的纯文本文件。
- SAM 或BAM 格式: 用来存储reads比对结果信息,SAM是可读的纯文本文件,然而BAM是SAM的二进制文本,一个压缩的可索引的reads比对文件。
- 染色体大小文件: 只有两列的纯文本文件,在“生物信息学文本处理大杂烩(一)”里已经讲过。hg19.chrom_24.sizes是人基因组hg19版本的size文件,是使用UCSC 的fetchChromSizes下载的。
- Fasta文件。
我主要使用的是比对后得到的BAM格式文件。
4. RSeQC软件进行质控检测
1. 使用bam_stat.py命令查看比对的总体情况
#命令说明
Usage: bam_stat.py [options]
Summarizing mapping statistics of a BAM or SAM file.
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-i INPUT_FILE, --input-file=INPUT_FILE
Alignment file in BAM or SAM format.
-q MAP_QUAL, --mapq=MAP_QUAL
Minimum mapping quality (phred scaled) to determine
"uniquely mapped" reads. default=30
#操作记录
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ bam_stat.py -i Scr.bam.sort
Load BAM file ... Done
#==================================================
#All numbers are READ count
#==================================================
Total records: 50976263
QC failed: 0
Optical/PCR duplicate: 0
Non primary hits 5208051 #表示多匹配位点
Unmapped reads: 1232377
mapq < mapq_cut (non-unique): 2464685
mapq >= mapq_cut (unique): 42071150
Read-1: 21096560
Read-2: 20974590
Reads map to '+': 21026875
Reads map to '-': 21044275
Non-splice reads: 18498057
Splice reads: 23573093
Reads mapped in proper pairs: 41139130
Proper-paired reads map to different chrom:0
确认代码:
bam_stat.py -i ${i}.bam.sort
2. 使用read_distribution.py命令查看基因组覆盖率
#命令说明:
Usage: read_distribution.py [options]
Check reads distribution over exon, intron, UTR, intergenic ... etc
The following reads will be skipped:
qc_failed
PCR duplicate
Unmapped
Non-primary (or secondary)
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-i INPUT_FILE, --input-file=INPUT_FILE
Alignment file in BAM or SAM format.
-r REF_GENE_MODEL, --refgene=REF_GENE_MODEL
Reference gene model in bed format.
#该命令需要输入两个文件
# -i为BAM或SAM文件
# -r为参考的bed文件
BED文件可以直接从RSeQC网站下载:
此次练习中下载了人的BED文件,测序数据为小鼠来源,错误!!
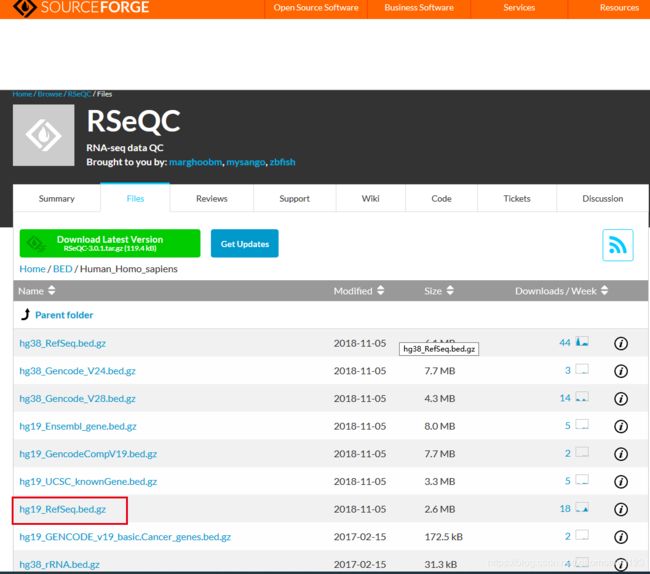 下载如下:
下载如下:
#使用wget下载,发现报错,暂未解决。
#在windows 10下面右键保存(2.6M)
#使用scp file [email protected]:~/dir 命令将文件上传至服务器
#使用gzip命令对文件解压缩,压缩文件不能被识别
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned/BED_file$ gzip -d hg19_RefSeq.bed.gz
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned/BED_file$ ls
hg19_RefSeq.bed
操作记录:
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ read_distribution.py -i ./Scr.bam.sort -r /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/BED_file/hg19_RefSeq.bed
processing /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/BED_file/hg19_RefSeq.bed ... Done
processing ./Scr.bam.sort ... Finished
Total Reads 44535835
Total Tags 75880491
Total Assigned Tags 37717256
=====================================================================
Group Total_bases Tag_count Tags/Kb
CDS_Exons 35271889 808827 22.93
5'UTR_Exons 13156148 236757 18.00
3'UTR_Exons 35031450 586598 16.74
Introns 1249039112 30136632 24.13
TSS_up_1kb 19566867 461946 23.61
TSS_up_5kb 88704854 1929741 21.75
TSS_up_10kb 163160166 3349520 20.53
TES_down_1kb 20819429 527166 25.32
TES_down_5kb 89844112 1590630 17.70
TES_down_10kb 160449429 2598922 16.20
=====================================================================
可以用一个饼图来表示,在生信技能树论坛里面还有人专门提问过。(有待进一步研究)
关于RSeQC输出结果的保存,可以使用定向写入【>】来保存。
#使用重定向>命令将read_distribution命令的输出结果定向到指定Log文件中
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ read_distribution.py -i ./Scr.bam.sort -r /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/BED_file/hg19_RefSeq.bed > Scr_distribution.log
processing /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/BED_file/hg19_RefSeq.bed ... Done
processing ./Scr.bam.sort ... Finished
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ ll
total 277G
-rw-rw-r-- 1 zexing zexing 1.2K 6月 4 13:05 Scr_distribution.log
#使用cat命令查看运行结果
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ cat Scr_distribution.log
Total Reads 44535835
Total Tags 75880491
Total Assigned Tags 37717256
=====================================================================
Group Total_bases Tag_count Tags/Kb
CDS_Exons 35271889 808827 22.93
5'UTR_Exons 13156148 236757 18.00
3'UTR_Exons 35031450 586598 16.74
Introns 1249039112 30136632 24.13
TSS_up_1kb 19566867 461946 23.61
TSS_up_5kb 88704854 1929741 21.75
TSS_up_10kb 163160166 3349520 20.53
TES_down_1kb 20819429 527166 25.32
TES_down_5kb 89844112 1590630 17.70
TES_down_10kb 160449429 2598922 16.20
=====================================================================
关于RSeQC的其他使用方法,参考文章:用RSeQC对比对后的转录组数据进行质控
3. 使用shell script对多个数据执行以上命令
关于脚本,参考文章:Linux学习笔记-学习shell脚本的使用(持续更新)
#脚本如下
#!/bin/bash
#program:
# This program is running RSeQC software command and saving the output results.
#History:
# 2020/06/04 zexing First release
for i in msh1 msh2 m3108 m3110 m3111 m3112 m3113 m3114
do
bam_stat.py -i ${i}.bam.sort > ${i}_bam_stat.log
read_distribution.py -i ${i}.bam.sort -r /f/xudonglab/zexing/projects/zhaoxiujuan/aligned/BED_file/hg19_RefSeq.bed > ${i}_distribution.log
done
#后台执行该命令
(base) zexing@DNA:~/projects/zhaoxiujuan/aligned$ nohup sh -x RSeQC.sh &
[1] 172776