蛋白质结构与功能的预测方法总结和资料汇总
蛋白质结构与功能的预测方法总结和资料汇总
“折叠(fold)”的概念
“折叠(fold)”是近年来蛋白质研究中应用较广的一个概念,它是介与二级和三级结构之间的蛋白质结构层次,它描述的是二级结构元素的混合组合方式。
*
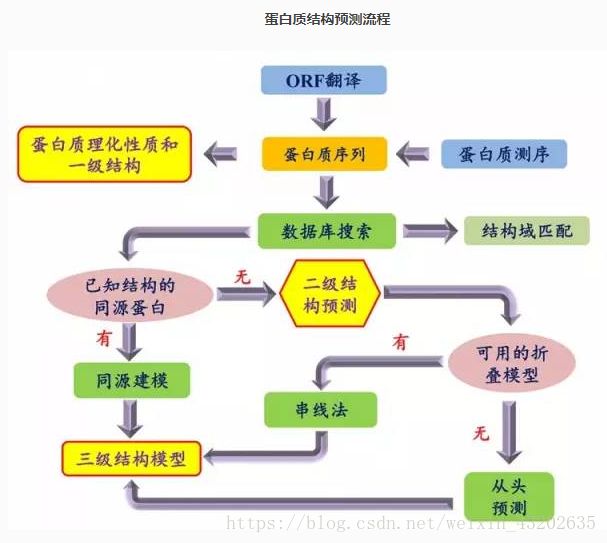
一。二级结构的预测方法介绍:
Chou-Fasman算法:
属于单序列预测方法,它是使用氨基酸物理化学数据中派生出来的规律来预测二级结构。
首先统计出20种氨基酸出现在α螺旋、β折叠和无规则卷曲中出现频率的大小,然后计算出每一种氨基酸在这几种构象中的构象参数Px。(构象参数值的大小反映了该种残基出现在某种构象中的倾向性的大小)
按照构象参数值的大小可以把氨基酸分为六个组:Ha(强螺旋形成者)、ha(螺旋形成者)、Ia(弱螺旋形成者)、ia(螺旋形成不敏感者)、ba(螺旋中断者)、Ba(强螺旋中断者)。
Chou和Fasman根据残基的倾向性因子提出二级结构预测的经验规则,要点是沿蛋白序列寻找二级结构的成核位点和终止位点。
这种方法可能能够正确反映蛋白质二级结构的形成过程,但预测成功率并不高,仅有50%左右
GOR算法:
属于单序列预测方法,因其作者Garnier, Osguthorpe和 Robson而得名。
这种方法是以信息论为基础的,也属于统计学方法的一种,GOR方法不仅考虑被预测位置本身氨基酸残基种类对该位置构象的影响,也考虑到相邻残基种类对该位置构象的影响。
这样使预测的成功率提高到 65% 左右。GOR方法的优点是物理意义清楚明确,数学表达严格,而且很容易写出相应的计算机程序,但缺点是表达式复杂。
多序列列线预测:
对序列进行多序列比对,并利用多序列比对的信息进行结构的预测。调查者可找到和未知序列相似的序列家族,然后假设序列家族中的同源区有同样的二级结构,预测不是基于一个序列而是一组序列中的所有序列的一致序列。
基于神经网络的序列预测:
利用神经网络的方法进行序列的预测,BP 网络即反馈式神经网络算法是目前二级结构预测应用最广的神经网络算法。
它通常是由三层相同的神经元构成的层状网络,使用反馈式学习规则,底层为输入层,中间为隐含层,顶层是输出层,信号在相邻各层间逐层传递,不相邻的各层间无联系,在学习过程中根据输入的一级结构和二级结构的关系的信息不断调整各单元之间的权重,最终目标是找到一种好的输入与输出的映象,并对未知二级结构的蛋白进行预测。
神经网络方法的优点是应用方便,获得结果较快较好,主要缺点是没有反映蛋白的物理和化学特性,而且利用大量的可调参数,使结果不易理解。
许多预测程序如PHD、PSIPRED等均结合利用了神经网络的计算方法。
基于已有知识的预测方法(knowledge based method):
这类预测方法包括Lim 和 Cohen 两种方法。
Lim 方法是一种物理化学的方法,它根据氨基酸残基的物理化学性质,包括:疏水性、亲水性、带电性以及体积大小等,并考虑残基之间的相互作用而制订出一套预测规则。对于小于50个氨基酸残基的肽链, Lim 方法的预测准确率可以达到73%.
Cohen 方法,它的提出当时是为了α/β蛋白的预测,基本原理是说:疏水性残基决定了二级结构的相对位置,螺旋亚单元或扩展单元是结构域的核心,α螺旋和β折叠组成了结构域。
混合方法(hybrid system method):
将以上几种方法选择性的混合使用,并调整他们之间使用的权重可以提高预测的准确率,目前预测准确率在70%以上的都是混合方法,其中,同源性比较方法、神经网络方法 和 GOR方法 应用最为广泛。
二。三级结构的预测:
同源性建模:
假设对已知结构的另一个蛋白质序列来排列一个蛋白质的序列,如果靶序列和已知结构序列在整个序列的全长有很高的相似性,在合理的信任度上,我们可以使用已知结构作为靶蛋白质的模版。
“串线(threading)”算法:
串线结构分析是试图把未知的氨基酸序列和各种已存在的三维结构相匹配,并评估序列折叠成那种结构的合适度。串线法最适用于折叠(fold)的识别,而不是模型的建立。
它是快速用未知序列的氨基酸侧链替换已知序列中的氨基酸位置。
Jones等首先从蛋白质结构数据库中挑选蛋白质结构建立折叠子数据库,以折叠子数据库中的折叠结构作为模板,将目标序列与这些模板一一匹配,通过计算打分函数值判断匹配程度,根据打分值给模板结构排序,其中打分最高的被认为是目标序列最可能采取的折叠结构。
Threading 方法的难点在于序列与折叠结构的匹配技术和打分函数的确定。(Jones等,1992)
*
三。蛋白质二级结构预测:
蛋白质二级结构的预测通常被认为是蛋白结构预测的第一步,是根据它们被预测的局部结构,对蛋白序列中的氨基酸进行分类。二级结构的预测方法通常分为多序列列线预测和单序列预测的方法。
由于单序列预测所提供的信息只是残基的顺序而没有其空间分布的信息,所以单序列预测的算法预测准确率并不高而且对于一些特殊结构,这些算法很难预测成功。
多序列列线预测和神经网络的应用大大提高了二级结构预测的准确度,通过对序列比对的预测可以明确的提供单一位点在三维结构上的信息。这样通常二级结构预测的准确率比单序列预测能够提高10%.许多方法据说可达到70%-77%,目前较为常用的几种方法有:PHD、PSIPRED、Jpred、 PREDATOR、PSA。其中最常用的是PHD。
PHD结合了许多神经网络的成果,每个结果都是根据局部序列上下文关系和整体蛋白质性质(蛋白质长度、氨基酸频率等)来预测残基的二级结构。那么,最终的预测是这些神经网络每个输出的算术平均值。 这种结合方案被称为陪审团决定法(jury decision)或者称为所有胜利者(winner-take-all)法。
PHD被认为是二级结构预测的标准。
‘
四。蛋白质三级结构预测:
蛋白质三维结构的预测方法通常包括:同源性建模和从头开始的预测方法。对数据库中已知结构的序列的比对是预测未知序列三级结构的主要方法,也即同源建模的方法。
通常对于同源建模的方法过程并非统一,但基本思路是一致的,基本包括如下几个步骤:
1.使用未知序列作为查询来搜索已知蛋白质结构。
2.产生未知序列和模版序列最可能的完整比对。
3.以模版结构骨架作为模型,建立蛋白质骨架模型。
4.在靶序列或者模版序列的有空位区域,使用环建模过程代替合适长度的片段。
5.给骨架模型加上侧链。
6.优化侧链的位置。
7.使用能量最小和已知的优化知识来优化结构。
在进行序列比对时,最容易使用 BLASTP 程序比对 NRL-3D 或 SCOP 数据库中的序列。如果发现超过100个碱基长度且有远高于40%序列相同率的匹配序列,则未知序列蛋白与该匹配序列蛋白将有非常相似的结构。在这种情况下,同源性建模在预测该未知蛋白精细结构方面会有非常大的作用。
同源性建模的成功的关键通常不是建模使用的软件或服务器,在设计与模版结构好的比对时的技巧更加重要。

*
五。常用蛋白序列和结构数据库:
数据库说明网址链接
PDB蛋白质三维结构http://www.rcsb.org/pdb
SWISS-PROT蛋白质序列数据库http://kr.expasy.org/sprot/
PIR蛋白质序列数据库http://pir.georgetown.edu/
OWL非冗余蛋白质序列http://www.bioinf.man.ac.uk/dbbrowser/OWL/
EMBL核酸序列数据库http://www.embl-heidelberg.de/
TrEMBLEMBL的翻译数据库http://kr.expasy.org/sprot/
GenBANK核酸序列数据库http://www.ncbi.nih.gov/Genbank/
PROSITE蛋白质功能位点http://kr.expasy.org/prosite/
SWISS-MODEL从序列模建结构http://www.expasy.org/swissmod/SWISS-MODEL.html
SWISS-3DIMAGE三维结构图示http://us.expasy.org/sw3d/
DSSP蛋白质二级结构参数http://www.cmbi.kun.nl/gv/dssp/
FSSP已知空间结构的蛋白质家族http://www.ebi.ac.uk/dali/fssp/fssp.html
SCOP蛋白质分类数据库http://scop.mrc-lmb.cam.ac.uk/scop/
CATH蛋白质分类数据库http://www.biochem.ucl.ac.uk/bsm/cath/
Pfam蛋白质家族和结构域http://pfam.wustl.edu/**
(一)蛋白质一级数据库
1、 SWISS-PROT 数据库
SWISS-PROT和PIR是国际上二个主要的蛋白质序列数据库,目前这二个数据库在EMBL和GenBank数据库上均建立了镜像 (mirror) 站点。
SWISS-PROT数据库包括了从EMBL翻译而来的蛋白质序列,这些序列经过检验和注释。该数据库主要由日内瓦大学医学生物化学系和欧洲生物信息学研究所(EBI)合作维护。SWISS-PROT的序列数量呈直线增长。
2、TrEMBL数据库:
SWISS-PROT的数据存在一个滞后问题,即把EMBL的DNA序列准确地翻译成蛋白质序列并进行注释需要时间。一大批含有开放阅读框(ORF) 的DNA序列尚未列入SWISS-PROT。为了解决这一问题,TrEMBL(Translated EMBL) 数据库被建立了起来。TrEMBL也是一个蛋白质数据库,它包括了所有EMBL库中的蛋白质编码区序列,提供了一个非常全面的蛋白质序列数据源,但这势必导致其注释质量的下降。
3、PIR数据库:
PIR数据库的数据最初是由美国国家生物医学研究基金会(National Biomedical Research Foundation, NBRF)收集的蛋白质序列,主要翻译自GenBank的DNA序列。
1988年,美国的NBRF、日本的JIPID(the Japanese International Protein Sequence Database日本国家蛋白质信息数据库)、德国的MIPS(Munich Information Centre for Protein Sequences摹尼黑蛋白质序列信息中心)合作,共同收集和维护PIR数据库。PIR根据注释程度(质量)分为4个等级。
4、 ExPASy数据库:
目前,瑞士生物信息学研究所(Swiss Institute of Bioinformatics, SIB)创建了蛋白质分析专家系统(Expert protein analysis system, ExPASy )。涵盖了上述所有的数据库。
网址:http://www.expasy.org
我国的北京大学生物信息中心(www.cbi.pku.edu.cn) 设立了ExPASy的镜像(Mirror)。
*
(二)蛋白质结构数据库
1、PDB数据库:
实验获得的三维蛋白质结构均贮存在蛋白质数据库PDB(Protein Data Bank)中。PDB是国际上主要的蛋白质结构数据库,虽然它没有蛋白质序列数据库那么庞大,但其增长速度很快。PDB贮存有由X射线和核磁共振(NMR)确定的结构数据。
2、NRL-3D 数据库:
NRL-3D(Naval Research Laboratory-3D)数据库提供了贮存在PDB库中蛋白质的序列,它可以进行与已知结构的蛋白质序列的比较。
3、HSSP数据库:
对来自PDB中每个已知三维结构的蛋白质序列进行多序列列线(multiple sequence alignment)同源性比较的结果,被贮存在HSSP(homology-derived second structures of proteins)数据库中。被列为同源的蛋白质序列很有可能具有相同的三维结构,HSSP因此根据同源性给出了SWISS-PROT数据库中所有蛋白质序列最有可能的三维结构。
4、 SCOP数据库:
要想了解对已知结构蛋白质进行等级分类的情况可利用SCOP(Structural classification of proteins)数据库,在该库中可以比较某一蛋白质与已知结构蛋白的结构相似性。
5、 CATH 数据库:
CATH(Class, Architecture, Topology and Homologous superfamily)是与SCOP类似的一个数据库。
六。 结构预测相关程序及数据库:
蛋白质预测分析网址集锦
物理性质预测
Compute PI/MW http://expaxy.hcuge.ch/ch2d/pi-tool.html
Peptidemasshttp://expaxy.hcuge.ch/sprot/peptide-mass.html TGREASE ftp://ftp.virginia.edu/pub/fasta/
SAPS http://ulrec3.unil.ch/software/SAPS_form.html
基于组成的蛋白质识别预测
AACompIdent http://expaxy.hcuge.ch … htmlAACompSim http://expaxy.hcuge.ch/ch2d/aacsim.html PROPSEARCH http://www.embl-heidelberg.de/prs.html
二级结构和折叠类预测
nnpredict http://www.cmpharm.ucsf.edu/~nomi/nnpredict
Predictprotein http://www.embl-heidel … protein/SOPMA http://www.ibcp.fr/predict.html
SSPRED http://www.embl-heidel … prd_info.html
特殊结构或结构预测
COILS http://ulrec3.unil.ch/ … ILS_form.html
MacStripe http://www.wi.mit.edu/ … acstripe.html
蛋白质预测在线分析常用软件集锦