16S预测细菌组表型-bugbase:革兰氏阴阳、生物膜、致病力、移动元件、氧气消耗等
基于16S OTU表预测细菌表型数据库,同时可进行组间差异分析
BugBase Predicts Organism Level Microbiome Phenotypes
Tonya Ward, Jake Larson, Jeremy Meulemans, Ben Hillmann, Joshua Lynch, Dimitri Sidiropoulos, John Spear, Greg Caporaso, Ran Blekhman, Rob Knight, Ryan Fink, Dan Knights
bioRxiv 133462; doi: https://doi.org/10.1101/133462
此文今天5月2日发布在预印本杂志bioRxiv上面,还没有正式发表。短短几个月己被引用3次,阅读2138次,全文下载789次。待正式发表发,必将成为本领域功能预测的又一神器。
居然有33条Tweets的转发和留言,看看大家的评价:

官网:https://bugbase.cs.umn.edu/

老司机直接Parse Data上传数据开分析,新人一定要先点下方Documentation,仔细阅读使用说明。
使用说明
BugBase是一款分析微生物组样品表型的工具,此网站可以基于OTU表和Mapping files,预测大量信息和比较,包括以下七方面:
- 革兰氏阳性 Gram Positive
- 革兰氏阴性 Gram Negative
- 生物膜形成 Biofilm Forming
- 致病潜力 Pathogenic Potential
- 移动元件含量 Mobile Element Containing
- 氧的利用 Oxygen Utilizing
- 氧化胁迫耐受 Oxidative Stress Tolerant
同时BugBase还会按组进行分类统计与可视化。
输入文件要求
OTU表
- BIOM 1.0格式
- 16S以GreenGenes 13.5 为Reference
- 宏基因组以IMG为参考
- 小于15 mb (本地版无限制)
Maping File
- 制表符分隔
- 第一行必须以#SampleID起始
- 第一行全为列标题
- 第一列必须为SampleID
- 只允许使用字母、数字、下划线和连字符
- 不允许包含空格,逗号、引号、括号
- 不要包含机密信息
其实就是符合QIIME的标准即可,如下图示例

分组列名:
指定分组信息,如上面的BODY_SITE,可产生如下结果:

还有很多其它功能,大家自己读帮助文档吧。
测试数据分析
- 可以点主页右边的Downloads,下载OTU表和Mapping file,
再到 Parse Data中上传这两个文件,Column Header添“BODY_SITE”,点Parse Data

很快生成了结果链接,打包下载所有结果。

本地化bugbase:安装和运行
对一直用服务器的小伙伴,肯定希望数据库有本地版,可以整合入自己的分析pipeline,随时搞,随便搞,一条命令搞定不求人。更不怕网站无法访问或无人维护。这个数据库还真可以本地方,满足muggle和geek的所有需求,安装代码如下:
源代码和数据库: https://github.com/knights-lab/BugBase
# 下载安装程序
cd ~/software
wget https://github.com/knights-lab/BugBase/archive/master.zip
mv master.zip BugBase.zip
unzip BugBase.zip
mv BugBase-master/ BugBase
# 此程序运行必须定义下面环境变量,根据实际目录修改
export BUGBASE_PATH=/home/user/software/BugBase
export PATH=$PATH:/home/user/software/BugBase/bin安装依赖R包并显示帮助
run.bugbase.r -h # 安装了所有依赖包
# 以上R包如果已经安装,此步可跳过
# 每次运行都会重复安装10多个包近半小时显示程序的使用帮助如下:内容较多,只显示前四个参数,完整信息见附录1。
所有依赖包列表和安装代码见附录2
Usage: /mnt/bai/yongxin/software/BugBase/bin/run.bugbase.r [options]
Options:
-i OTUTABLE, --otutable=OTUTABLE
otu table to plot [default NULL]
-c MAPCOLUMN, --mapcolumn=MAPCOLUMN
column of mapping file to plot [default NULL]
-m MAPPINGFILE, --mappingfile=MAPPINGFILE
mapping file to plot [default NULL]
-o OUTPUT, --output=OUTPUT
output directory [default .]
运行演示数据
# 运行演示数据
run.bugbase.r -i $BUGBASE_PATH/doc/data/HMP_s15.txt -m $BUGBASE_PATH/doc/data/HMP_map.txt -c HMPBODYSUBSITE -o output运行中会显示运行内容如下
[1] "Loading Inputs..."
[1] "16S copy number normalizing OTU table..."
[1] "Predicting phenotypes..."
[1] "313 OTUs from the input table matched the 203452 available database OTUs"
[1] "Plotting thresholds..."
[1] "Plotting predictions..."
[1] "Plotting OTU contributions..."
[1] "BugBase analysis complete"输入参数文件详解
- OTU表
-i$BUGBASE_PATH/doc/data/HMP_s15.txt
less -S $BUGBASE_PATH/doc/data/HMP_s15.txt
# Constructed from biom file
#OTU ID SRS024625.570280 SRS018102.573993
963239 0.0 0.0 0.0 0.0 0.0 0.0
4431292 28.0 0.0 0.0 16.0 1.0 12.0
4480529 0.0 0.0 0.0 10.0 3.0 3.0 - mapping file
-m$BUGBASE_PATH/doc/data/HMP_map.txt
sed -i 's/^M/\n/g' BUGBASE_PATH/doc/data/HMP_map.txt
less -S $BUGBASE_PATH/doc/data/HMP_map.txt
#SampleID BarcodeSequence SAMP_COLLECT_DEVICE TITLE RUN_PREFIX AGE COMMON_NAME BODY_SITE
SRS024568.571681 TCAGAATACGTTC stool_specimen_collection_kit HMP_production_phase_2715 SRR040712
SRS013177.574389 TCAGAGTCGAC stool_specimen_collection_kit HMP_production_phase_199 SRR040857
SRS020413.572892 TCAGAACCGGATAC stool_specimen_collection_kit HMP_production_phase_1681 SRR040994 - 分组列名
-cHMPBODYSUBSITE
输出文件详解
输出结果在线分析直接生成压缩包下载。本地在Ouput目录中有四个目录:
normalized_otus目录中有16s_normalized_otus.txt文件,为标准化的OTU表
less -S output/normalized_otus/16s_normalized_otus.txt
# 文件如下:
SRS024568.571681 SRS013177.574389 SRS020413.572892
963239 0 0 0 0 0 0 0 0 0
4431292 0 0 0 2 11.75 3 3 7 0
4480529 0 0 3 0 8 3 27 0 0
4345640 0 0 0 0 0 0 0 0 0
4372091 0 0 0 0 0 0 0 0 0otu_contributions目录中
主要有9种表型或功能预测结果表contributing_otus.txt,和9种表型按实验组比较的结果堆叠柱状图和物种颜色方案图例PDF版
less -S output/otu_contributions/contributing_otus.txt
# 预测结果前4行内容如下
Aerobic Anaerobic Contains_Mobile_Elements Facultatively_Anaerobic Forms_Biofilms Gram_Negative Gram_Positive Potentially_Pathogenic Stress_Tolerant
963239 FALSE TRUE TRUE FALSE FALSE FALSE TRUE FALSE TRUE
4431292 FALSE TRUE TRUE FALSE FALSE FALSE TRUE TRUE TRUE
4480529 FALSE TRUE TRUE FALSE FALSE FALSE TRUE FALSE TRUE
图0. 注释物种门水平图例

图1. 各组需氧菌相对丰度

图2 各组厌氧菌相对丰度
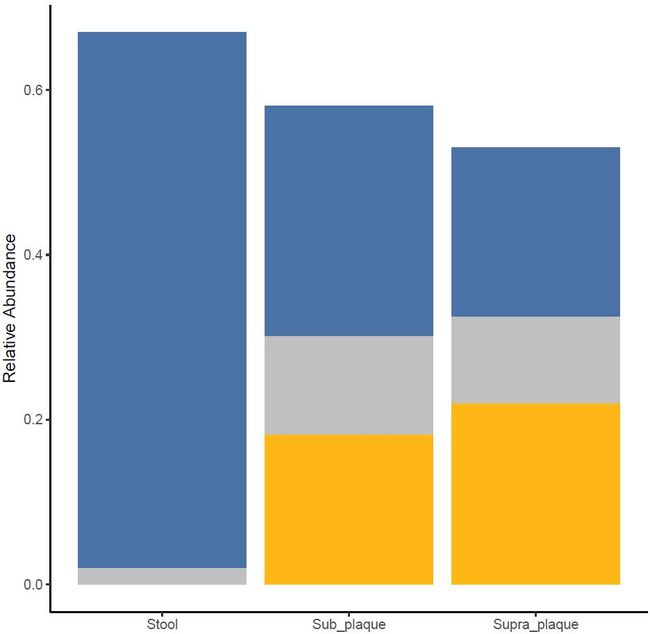
图3. 革兰氏阴性菌相对丰度
一共有9种,不再一一列举
predicted_phenotypes目录中
主要有9种表型或功能预测结果表predictions.txt,和9种表型按实验组比较箱线图,和相关组间统计信息。
less -S output/predicted_phenotypes/predictions.txt
Aerobic Anaerobic Contains_Mobile_Elements Facultatively_Anaerobic Forms_Biofilms Gram_Negative Gram_Posi
SRS024568.571681 0 0.999124411409892 0.219286298011441 0 0 0.782173016305405 0.2178269
SRS013177.574389 0 1 0.185520182485371 0 0 0.82138252504215 0.17861747495785 0
SRS020413.572892 0 0.99738106419102 0.347650182422425 0.00180616262688292 0 0.687353249286566
图4. 箱线图展示9种表型中的移动元件含量
thresholds目录中包含分析使用的阈值thresholds_used.txt和不同阈值下的结果variances.txt数据,以及9表型在不同阈值下相对丰度变化

图5. 折线图展示9种表型中的生物膜形成菌在不同阈值下相对丰度变化
Reference
- BugBase Predicts Organism Level Microbiome Phenotypes
Tonya Ward, Jake Larson, Jeremy Meulemans, Ben Hillmann, Joshua Lynch, Dimitri Sidiropoulos, John Spear, Greg Caporaso, Ran Blekhman, Rob Knight, Ryan Fink, Dan Knights
bioRxiv 133462; doi: https://doi.org/10.1101/133462
附录1. 程序参数详解
Usage: /mnt/bai/yongxin/software/BugBase/bin/run.bugbase.r [options]
Options:
-v, --verbose
Print extra output [default]
-i OTUTABLE, --otutable=OTUTABLE
otu table to plot [default NULL]
-c MAPCOLUMN, --mapcolumn=MAPCOLUMN
column of mapping file to plot [default NULL]
-m MAPPINGFILE, --mappingfile=MAPPINGFILE
mapping file to plot [default NULL]
-o OUTPUT, --output=OUTPUT
output directory [default .]
-t TAXALEVEL, --taxalevel=TAXALEVEL
taxa level to plot otu contributions by, default is
2 (phylum) [default NULL]
-p PHENOTYPE, --phenotype=PHENOTYPE
specific traits (phenotypes) to predict, separated by
commas, no spaces [default NULL]
-x, --predict
only output the prediction table, do not make plots
[default FALSE]
-T THRESHOLD, --threshold=THRESHOLD
threshold to use, must be between 0 and 1
[default NULL]
-g GROUPS, --groups=GROUPS
treatment groups of samples, separated by commas, no spaces
[default NULL]
-u USERTABLE, --usertable=USERTABLE
user define trait table, absolute file path required [
default NULL]
-z, --continuous
plot continuous data [default FALSE]
-k, --kegg
use kegg pathway table [default FALSE]
-C, --cov
use coefficient of variance instead of variance [default FALSE]
-l, --clr_trans
use centered log-ratio transformation instead of relative abundance [default FALSE]
-a, --all
plot all samples without a mapping file (this outputs no
statistics) [default FALSE]
-w, --shotgun
Data is metagenomic shotgun data
(picked against RefSeq database) [default FALSE]
-h, --help
Show this help message and exit附录2. 安装所有依赖R包并测试
# 运行R
R
# 安装13个依赖包,环境为Ubuntu16.04 + R3.4.1,祝你成功
source("https://bioconductor.org/biocLite.R")
biocLite(c("optparse", "beeswarm", "RColorBrewer", "reshape2", "plyr", "grid", "gridExtra", "ggplot2", "RJSONIO", "biom", "Matrix", "labeling", "digest"))
# 测试是否安装成功
library("optparse")
library("beeswarm")
library("RColorBrewer")
library("reshape2")
library("plyr")
library("grid")
library("gridExtra")
library("ggplot2")
library("RJSONIO")
library("biom")
library("Matrix")
library("labeling")
library("digest")
q()附录3. 常见错误
- otu表格式错误
[1] "Loading Inputs..."
Error in load.inputs(otu_table, map, mapcolumn, groups) :
Error: otu table must be either .txt or .biom (json)
Execution halted解决方法:要求扩展名符合要求,如文本表必须为.txt结尾,更正即可
- 输出目录己存在
Error: Output directory already exists
Execution halted程序防止覆盖结果,手动删除输出目录即可,或换个不存在的输出目录
附录4. 革兰氏细菌分类解决思路
百度搜索“革兰氏 分类 数据库”没有结果
http://mp.weixin.qq.com/s/uWdJy_i-8Wu_uwifyPsqDg 文章中提到了细菌分类,但作者说是自己整理的数据库,且不可以给我
google搜索“Gram stain database”
主要相关结果:
- https://www.biostars.org/p/201794/ biostar和我同样的问题,大家给了如下多条建议
- http://www.globalrph.com/bacterial-strains-background.htm 临床最终参考细菌数据库,其中革兰氏阴、阳;只有一些名字,多个网页,需要整理
- https://www.biostars.org/p/154240/ 此文给出了wiki和分类手册的链接
- https://en.wikipedia.org/wiki/Category:Gram-positive_bacteria 有列表链接,比较好整理 https://en.wikipedia.org/wiki/Category:Gram-negative_bacteria
- https://archive.org/details/bergeysmanualofd1957amer 1957年分类手册,太老了吧
- https://www.biostars.org/p/126384/#126392 NCBI有文件lproks_0.txt,染色为菌的一种表型
但完全没能快速找到针对性的整理数据库或软件。后来在在群中提问:“有基于16S鉴定革兰氏分类的数据库吗?”,两个群里都有人回答了bugbase。使用发现专门解决我的问题,只是没有发表的软件,怪不得查不到,但也显示了大家知识的力量。一句话解决了可能几天都无法解决的问题。
猜你喜欢
- 一文读懂:1微生物组 2进化树 3预测群落功能
- 热文:1图表规范 2DNA提取 3 实验vs分析
- 必备技能:1提问 2搜索 3Endnote
- 文献阅读 1热心肠 2SemanticScholar 3geenmedical
- 扩增子分析:1图表解读 2分析流程 3统计绘图 4群落功能 5进化树
- 科研团队经验:1云笔记 2云协作 3公众号
- 系列教程:1Biostar 2微生物组 3宏基因组
- 生物科普 1肠道细菌 2生命大跃进 3细胞的暗战 4人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外100+ PI,800+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习扩增子、宏基因组科研思路和分析实战,关注“宏基因组”

点击阅读原文,跳转最新文章目录阅读
https://mp.weixin.qq.com/s/5jQspEvH5_4Xmart22gjMA