
如果您的组织类型不存在分类器我们的仓库中,或者数据中不包含您期望的细胞类型,那么您需要生成自己的分类器。
训练分类器的第一步是加载单细胞数据。
因为Garnett 建立在 Monocle上,所以Garnett 的数据保存在CellDataSet (CDS)类的对象中。这个类派生自Bioconductor ExpressionSet类,它为那些分析过生物微阵列实验的人提供了一个常见的接口。Monocle提供了关于如何生成输入cds的详细文档here。
例如,Garnett包含一个来自PBMC 10x V1表达式数据的小数据集.
library(monocle3)
library(garnett)
# load in the data
# NOTE: the 'system.file' file name is only necessary to read in
# included package data
#
mat <- Matrix::readMM(system.file("extdata", "exprs_sparse.mtx", package = "garnett"))
fdata <- read.table(system.file("extdata", "fdata.txt", package = "garnett"))
pdata <- read.table(system.file("extdata", "pdata.txt", package = "garnett"),
sep="\t")
row.names(mat) <- row.names(fdata)
colnames(mat) <- row.names(pdata)
# create a new CDS object
#pd <- new("AnnotatedDataFrame", data = pdata)
#fd <- new("AnnotatedDataFrame", data = fdata)
pbmc_cds <- new_cell_data_set(as(as.matrix(mat), 'sparseMatrix'),
cell_metadata = pdata,
gene_metadata = fdata)
# generate size factors for normalization later
#pbmc_cds <- estimateSizeFactors(pbmc_cds)#
pbmc_cds = preprocess_cds(pbmc_cds, num_dim = 100)
构造标记(marker )文件
除了表达数据之外,您需要的第二个主要输入是一个标记文件(marker file)。标记文件包含以易于阅读的文本格式编写的单元类型定义列表。细胞类型定义告诉Garnett如何选择细胞来训练模型。每个细胞类型定义以“>”符号和细胞类型名称开头,后面是一系列带有定义信息的行。定义行以关键字和':'开头,条目用逗号分隔。
一个简单有效的例子:
>B cells
expressed: CD19, MS4A1
>T cells
expressed: CD3D
有几种方法可以在Garnett标记文件格式中定义细胞类型。通常,每个细胞的定义可以包含三个主要组件。只需要第一个组件。
细胞类型的第一个也是最重要的规范是它的表达式。Garnett提供了几种指定标记基因的选项,详情如下。
Format
expressed: gene1, gene2
not expressed: gene1, gene2
这是为你的细胞类型指定标记基因的默认方法。在使用这个规范时,Garnett为每个细胞计算一个标记分数,包括潜在的泄漏(leakage)、总体表达水平和read 深度。expressed:标记应该特定于所定义的细胞类型。
| Format | Example |
|---|---|
| expressed above: gene1 value, gene2 value | expressed above: MYOD1 4.2, MYH3 700 |
| expressed below: gene1 value, gene2 value | expressed below: PAX6 20, PAX3 4 |
| expressed between: gene1 value1 value2, gene2 value1 value2 | expressed between: PAX6 10 20, PAX3 4 100 |
这是指定expression的另一种方法,如果您知道某个基因的确切表达范围,这种方法会很有用。但是,通常我们不建议使用这些规范,因为它们不会考虑每个细胞中的read深度和总体表达。数据值与输入数据的单位相同。
定义元数据
除了表达式信息之外,您还可以使用元数据进一步细化细胞类型定义。这也是指定数据中所期望的任何子类型的地方。
| Format | Example |
|---|---|
| subtype of: celltype | subtype of: T cells |
| custom meta data: attribute1, attribute2 | tissue: spleen, thymus |
subtype of:允许您在定义文件中指定细胞类型是另一个细胞类型的子类型。
custom meta data:规范允许您为细胞类型提供任何进一步的元数据需求。CDS对象的pData表中的任何列都可以用作元数据规范。在上面的示例中,pData表中有一个名为“tissue”的列。
提供你的证据
最后,我们强烈建议您记录如何选择标记定义。为了便于跟踪,我们提供了一个额外的规范—references:—它将存储每种类型的引用信息。添加一组url或DOIs,它们将包含在分类器中。有关访问此信息的函数,请参见此处。
添加注释
与R代码类似,我们已经包含了一个注释字符#,这样您就可以在您的标记文件中添加注释/注释。任何在同一条线上的#之后的内容都会被忽略。
所以一个完整的例子是这样的:
>B cells
expressed: CD19, MS4A1
expressed above: CD79A 10
references: https://www.abcam.com/primary-antibodies/b-cells-basic-immunophenotyping,
10.3109/07420528.2013.775654
>T cells
expressed: CD3D
sample: blood # A meta data specification
>Helper T cells
expressed: CD4
subtype of: T cells
references: https://www.ncbi.nlm.nih.gov/pubmed/?term=1200072
目前,标记文件不能有回车(\r),如果您在某些Windows文本编辑器中生成标记文件,就会自动包括回车。相反,您必须使用换行字符(\n)。有许多方法可以在Windows上替换回车,例如使用notepad++,您可以使用replace、choose extended和replace all \r with \n。我打算尽快以更自动化的方式解决这个问题。
检查标记
因为定义标记文件通常是过程中最困难的部分,所以Garnett提供了一些函数来检查标记是否能够正常工作。相关的两个函数是check_marker和plot_marker。check_marker生成关于标记的信息表,plot_marker绘制最相关的信息。
除了包含的小数据集之外,我们还在包中包含了两个示例标记文件:pbmc_bad_markers.txt
在生成您的marker_check data.frame 之后,您可以使用我们内建的绘图函数plot_marker来查看结果。
library(org.Hs.eg.db)
marker_file_path <- system.file("extdata", "pbmc_bad_markers.txt",
package = "garnett")
marker_check <- check_markers(pbmc_cds, marker_file_path,
db=org.Hs.eg.db,
cds_gene_id_type = "SYMBOL",
marker_file_gene_id_type ="SYMBOL")
plot_markers(marker_check)
-
db:dbis a required argument for a Bioconductor AnnotationDb-class package used for converting gene IDs. For example, for humans use org.Hs.eg.db. See available packages at the Bioconductor website. Load your chosen db usinglibrary(db). If your species does not have an AnnotationDb-class package, see here. -
cds_gene_id_type: This argument tells Garnett the format of the gene IDs in your CDS object. It should be one of the values incolumns(db). The default is "ENSEMBL". -
marker_file_gene_id_type: Similarly to above, this argument tells Garnett the format of the gene IDs in your marker file.
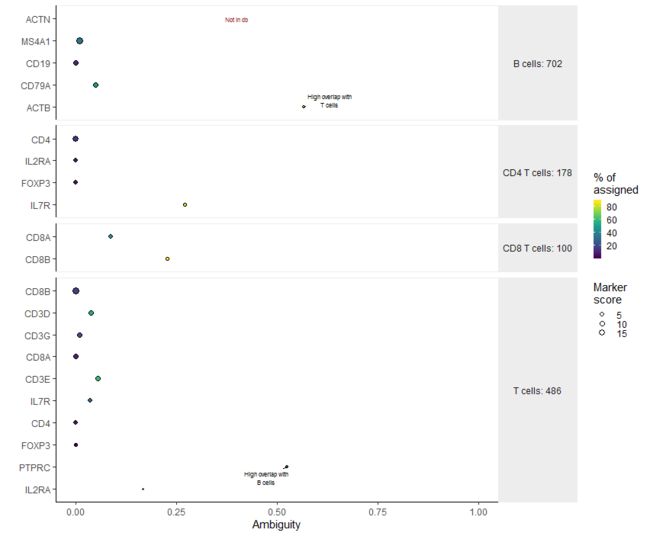
这个标记图提供了一些关于所选标记是否正确的关键信息。首先,红色标记“not in db”让我们知道标记ACTN在org.Hs.eg.db注释中没有作为“SYMBOL”出现。在本例中,它是一个打印错误。接下来,x轴显示每个标记的模糊度评分—当包含该标记时,测量有多少个cell接受了模糊标签—在本例中,ACTB和PTPRC具有很高的模糊度,应该排除。
check_marker输出的值和plot_marker绘制的值是分类器可以选择的cell 数量的估计值。然而,它使用启发式快速找到候选细胞,并不能完全匹配标记所选择的细胞。请使用这些数字作为相对的度量,而不是训练集的绝对表示。
关于歧义分数的进一步说明:歧义分数是当一个标记被包含在标记文件中时,一个标记被标记为“歧义”的cell 的分数。然而,一个高模糊度的分数并不一定意味着一个给定的标记是不具体的。这可能意味着一个不同的标记是罪魁祸首,但该标记也提名了许多其他未标记的细胞(高提名率)。在决定排除哪个标记之前,请仔细查看歧义度高的标记和最模糊的cell类型marker。
在制作标记图之后,您可能想要修改标记文件。在我们的示例中,我们将删除ACTN、ACTB和PTPRC以得到最终的“pbmc_test”。txt的标记文件。
默认情况下,Garnett 将基因id转换成ENSEMBL用于分类器。这使得分类器是可移植的,这样它们就可以对未来可能具有不同基因id的数据集进行分类。然而,对于某些生物体,ENSEMBL id要么不可用,要么不常用。在这些情况下,可以在train_cell_classifier和check_marker中使用参数classifier_gene_id_type来指定不同的ID类型。您选择的值将与分类器一起存储,因此在对未来的数据集进行分类时不需要再次指定它。
训练分类器
现在是训练分类器的时候了。参数应该与check_marker的参数非常接近。下面我将从默认值更改的一个参数是num_unknown参数。这告诉Garnett 应该比较多少个外群细胞。默认值是500,但是在这个只有很少cell的数据集中,我们需要更少的cell。
library(org.Hs.eg.db)
set.seed(260)
marker_file_path <- system.file("extdata", "pbmc_test.txt",
package = "garnett")
pbmc_classifier <- train_cell_classifier(cds = pbmc_cds,
marker_file = marker_file_path,
db=org.Hs.eg.db,
cds_gene_id_type = "SYMBOL",
num_unknown = 50,
marker_file_gene_id_type = "SYMBOL")
head(pData(pbmc_cds))
> head(pData(pbmc_cds))
DataFrame with 6 rows and 4 columns
tsne_1 tsne_2
AAGCACTGCACACA-1 3.8403149909359 12.0841914129204
GGCTCACTGGTCTA-1 9.97096226657347 3.50539308651821
AGCACTGATATCTC-1 3.45952940410281 4.93527280576176
ACACGTGATATTCC-1 1.74394947394641 7.78267061846286
ATATGCCTCTGCAA-1 5.78344829514223 8.55889827553495
TGACGAACCTATTC-1 10.7928530485958 10.5852739146963
Size_Factor FACS_type
AAGCACTGCACACA-1 0.559181445161514 B cells
GGCTCACTGGTCTA-1 0.515934033527584 B cells
AGCACTGATATCTC-1 0.698028398302026 B cells
ACACGTGATATTCC-1 0.815631008885519 B cells
ATATGCCTCTGCAA-1 1.11532798424345 B cells
TGACGAACCTATTC-1 0.649469901028841 B cells
运行train_cell_classifier之后,类型为“garnett_classifier”的输出对象包含对细胞进行分类所需的所有信息。
查看分类基因
Garnett 分类是使用多项弹性-网络回归训练(multinomial elastic-net regression)。这意味着选择某些基因作为区分细胞类型的相关基因。所选择的基因可能是有趣的,所以Garnett 包含了一个访问所选择基因的功能。注意:Garnett 没有对输入标记进行正则化,所以无论如何,它们都会被包含在分类器中。
我们用来查看相关基因的函数是get_feature_genes。参数是分类器,您想查看哪个节点(如果您的树是分层的)—使用“root”作为顶部节点,使用父细胞类型名称作为其他节点,使用db作为您的物种。如果设置convert_ids = TRUE,该函数将自动将基因id转换为符号。
feature_genes <- get_feature_genes(pbmc_classifier,
node = "root",
db = org.Hs.eg.db)
head(feature_genes)
B cells T cells Unknown
(Intercept) -4.44471924 2.590206989 1.85451225
ENSG00000187608 -0.10903325 -0.199658082 0.30869133
ENSG00000157873 0.06411921 -0.295797377 0.23167817
ENSG00000157191 -0.07677221 -0.155893113 0.23266532
ENSG00000173436 0.02649831 0.089393639 -0.11589195
ENSG00000117318 0.07400983 -0.009415156 -0.06459467
Viewing references
我们在上面解释了如何在标记文件中包含关于如何选择标记的文档。为了获取这些信息—查看如何为已经训练好的分类器选择标记—使用函数get_classifier_references。除了分类器之外,还有一个额外的可选参数,称为cell_type。如果传递细胞类型的名称,则仅打印该单元类型的引用,否则将全部打印。
get_classifier_references(pbmc_classifier )
$`B cells`
[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=23688120"
[2] "https://www.ncbi.nlm.nih.gov/pubmed/?term=21149806"
$`T cells`
[1] "https://www.ncbi.nlm.nih.gov/pubmed/?term=1534551"
提交一个分类器
我们鼓励你提交你的高质量的分类器给我们,这样我们可以使他们对社区可用。为此,打开一个特刊并在Garnett github存储库中填写表单。点击这里,点击“New issue”按钮开始!
应用您的分类器
单细胞转录组数据分析||Garnett细胞类型注释工具