前情提要:实现Stingtie与DEseq2的无缝连接
RNA-seq数据分析下文是将StringTie产生的数据利用DESeq2进行处理。
DEseq2是转录组R统计分析的重要步骤,有广泛的适用性,可以与大多数的上游软件进行连接,在前文章中stringtie已经生成了数据:
gene_count_matrix.csv,
PHENO_DATA:geuvadis_phenodata.csv,
原始数据:ftp://ftp.ccb.jhu.edu/pub/RNAseq_protocol
数据转化
我们就以此为例进行分析。首先是将生成的数据转化为DESeq2所需要的文件,colData,和countData:
> countData <- as.matrix(read.csv("gene_count_matrix.csv", row.names="gene_id"))
> colData <- read.csv("geuvadis_phenodata.csv", row.names=1)
-
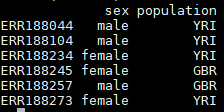
colData:是给每个样本进行标签化,如图所示分别有sex和population两个标签。下图为colData的内容:
 countData
countData -
countData:countData是反映了基因的表达量。MSTRG*为基因名,ERR*为样本名,数字代表样本每个基因的表达量。下图为countData的格式:
 colData
colData
进入R,并载入DESeq2的包
> R
>library("DESeq2")
由countData,colData生成差异表达数据:
> dds <- DESeqDataSetFromMatrix(countData = countData, colData = colData, design = ~ population)
生成的dds文件的内容,dds是一个类,其对生成表格的各个方面进行了描述:
class: DESeqDataSet
dim: 728 12
metadata(1): version
assays(4): counts mu H cooks
rownames(728): MSTRG.5 MSTRG.1 ... MSTRG.589 MSTRG.662
rowData names(22): baseMean baseVar ... deviance maxCooks
colnames(12): ERR188044 ERR188104 ... ERR188454 ERR204916
colData names(3): sex population sizeFactor
表达量的筛选
在countData文件里,将每个基因所有的表达量加起来大于10的留下来,并输入到dds文件中。
> keep <- rowSums(counts(dds)) >= 10
> dds <- dds[keep,]
将dds进行DESeq处理
resultsNames是输出默认的比对的顺序。在Fold Change的计算中需要比对的顺序,在这里默认的比对顺序为:
> dds <- DESeq(dds)
> resultsNames(dds)
DESeq处理内容如下:
estimating dispersions
found already estimated dispersions, replacing these
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
输出的内容:
[1] "Intercept" "population_YRI_vs_GBR"
pvalue与adjust pvalue(padj)的转换
通过规定alpha的值,来进行转换,值越小,筛选越严格,默认的alpha值为0.1,这里可以用res05输出alpha的值为0.05时的adjust pvalue。
contrast规定Fold Change的比对顺序。
> res <- results(dds)
> res <- results(dds, alpha=0.1,)
> contrast=c("population","GBR","YRI"))
> res05 <- results(dds, alpha=0.05)
生成res内容如下:
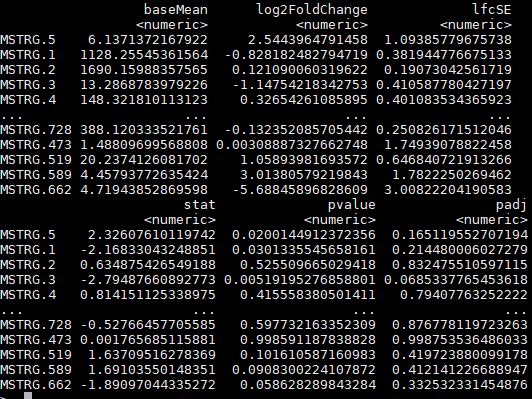
adjust pvalue进行排序
将pvalue或者是padj进行排序,方便进一步的筛选的有意义的差异表达。
# 对res里padj的大小进行排序;
> resOrdered <- res[order(res$padj),]
#对res进行了描述;
> summary(res)
#查看resOrdered的前五行的信息
> head(resOrdered)
#查看p-values>0.1的基因个数;
> sum(res$padj < 0.1, na.rm=TRUE)
#查看p-values>0.05的基因个数;
> sum(res05$padj < 0.05, na.rm=TRUE)
#对padj进行排序。
> res05Ordered <- res05[order(res05$padj),]
开始进行作图
1. MA-plot
利用一下命令可以做出相应的MA图:
plotMA(res, ylim=c(-2,2))
MA-plot表示了基因丰度和表达变化之间的关系,横坐标为基因表达量,纵坐标为基因的差异表达的大小。每个点是代表转录本。红色的点为要取的点,黑色的点为不好的点。生成的图标如下:

[图片上传中...(plot Counts.png-2283c9-1542618060107-0)]
2. Plot counts
此图是为了表达在不同的条件下基因的表达量的差异,横坐标为不同的条件,纵坐标为表达量,如下图所示,两组条件下,基因的表达量差异性比较大

将结果以CSV的格式文件输出。
write.csv(as.data.frame(resOrdered),
file="population_GBR_results.csv")
官方命令附件:https://bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.R