前言
NormqPcR这个包用于对qPCR数据进行均一化(normalization,这里需要注意的是,这里的normalization只是使用内参来校正目标基因的表达的意思,与统计学中的归一化(Normalization),中心化(center),标准化(scales)之类的术语没有关系)。这篇笔记主要是翻译了这个包的英文文档,本文档以一个小型案例(vignette)来演示了这个包的主要功能,笔记主要内容如下所示:
- 合并技术重复(technical replicates)(了解即可);
- 处理低Cq值(undetermined values)(了解即可);
- 计算用于均一化的最佳管家基因(housekeeping genes)(这个最重要,其余的很多函数其实了解即可,这一部分要了解geNorm和NormFinder的算法原理);
- 使用管家基因来对数据进行均一化(normalization)(了解)。
注:在这篇笔记中,管家基因(housekeeping genes)与内参(reference)指的是同一个意思。
合并技术重复
我们先使用ReadqPCR包来导入原始数据,这个原始数据中含有技术重复,其后缀都是_TechRep.n,其中n表示总的重复数。读取数据过程如下所示:
# BiocManager::install("ReadqPCR")
# BiocManager::install("NormqPCR")
# input raw data for qPCR
library(NormqPCR)
library(ReadqPCR)
path <- system.file("exData", package = "ReadqPCR")
qPCR.example.techReps <- file.path(path, "qPCR.techReps.txt")
qPCRBatch.qPCR.techReps <- read.qPCR(qPCR.example.techReps)
rownames(exprs(qPCRBatch.qPCR.techReps))[1:8]
计算结果如下所示:
> rownames(exprs(qPCRBatch.qPCR.techReps))[1:8]
[1] "gene_aj_TechReps.1" "gene_aj_TechReps.2" "gene_al_TechReps.1"
[4] "gene_al_TechReps.2" "gene_ax_TechReps.1" "gene_ax_TechReps.2"
[7] "gene_bo_TechReps.1" "gene_bo_TechReps.2"
上面是读取数据的一些信息,从中我们可以发现,里面有技术重复,例如gene_aj_TechReps.1与gene_aj_TechReps.2就是做了2个技术重复。
在我们计算之前,我们还要计算技术重复Cq的算术平均值(arithmetic mean),这一步我们使用combineTechReps()函数来计算一下,这个函数会生成一个qPCRBatch对象,这样就计算出了技术重复的算术平均值,如下所示:
# Cal arithmetic mean of the raw qPCR Cq value
# combineTechReps function will return a new qPCRBatch object
combinedTechReps <- combineTechReps(qPCRBatch.qPCR.techReps)
combinedTechReps
exprs(qPCRBatch.qPCR.techReps)
exprs(combinedTechReps)
apply(exprs(qPCRBatch.qPCR.techReps)[1:2,],2,mean)
结果如下所示:
> combinedTechReps <- combineTechReps(qPCRBatch.qPCR.techReps)
> combinedTechReps
qPCRBatch (storageMode: lockedEnvironment)
assayData: 8 features, 4 samples
element names: exprs
protocolData: none
phenoData
sampleNames: four one three two
varLabels: sample
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation:
> exprs(qPCRBatch.qPCR.techReps)
four one three two
gene_aj_TechReps.1 18.02512 20.72960 21.87109 17.51878
gene_aj_TechReps.2 17.59243 19.58196 21.71061 18.40029
gene_al_TechReps.1 20.30233 22.23137 19.30596 21.32223
gene_al_TechReps.2 20.56011 21.66608 20.07169 19.53765
gene_ax_TechReps.1 20.67044 19.09129 21.90034 21.15897
gene_ax_TechReps.2 20.44186 18.22294 21.28660 21.31048
gene_bo_TechReps.1 20.62939 18.77342 19.70848 21.08782
gene_bo_TechReps.2 20.70826 17.95030 19.53260 21.53267
gene_cp_TechReps.1 23.50768 20.39530 22.06632 22.99578
gene_cp_TechReps.2 22.73511 19.64446 20.74793 22.34119
gene_dx_TechReps.1 21.16744 19.57131 21.62546 21.65467
gene_dx_TechReps.2 21.36217 20.21180 21.41619 20.73930
gene_ei_TechReps.1 19.90058 19.78343 20.26527 19.83862
gene_ei_TechReps.2 19.67431 20.97861 19.82184 19.16033
gene_jy_TechReps.1 21.60261 19.23546 18.22053 22.21470
gene_jy_TechReps.2 21.04445 18.75998 19.31650 20.94050
> exprs(combinedTechReps)
four one three two
gene_aj 17.80877 20.15578 21.79085 17.95954
gene_al 20.43122 21.94873 19.68883 20.42994
gene_ax 20.55615 18.65711 21.59347 21.23473
gene_bo 20.66882 18.36186 19.62054 21.31024
gene_cp 23.12139 20.01988 21.40713 22.66848
gene_dx 21.26481 19.89156 21.52082 21.19699
gene_ei 19.78745 20.38102 20.04355 19.49948
gene_jy 21.32353 18.99772 18.76851 21.57760
> apply(exprs(qPCRBatch.qPCR.techReps)[1:2,],2,mean)
four one three two
17.80877 20.15578 21.79085 17.95954
从上面我们可以看到:
- 做了2个技术重复;
- 使用
combineTechReps()函数计算了技术重复的算术平均值; - 又用
apply()函数核对了一下,没问题。
处理未检测值
在qPCR实验中,当Cq值超过一定阈值后,qPCR仪有可能会将这个读数标记为Undetermined,也就是数值太小(Cq值越大,表示信号越弱),在qPCRBatch对象中,这个值就会被标记为NA。不同的人有不同的处理策略,常见的处理策略有两种。
第一种就是将那些大于38的Cq值认为是无法检测值,并将其标记为40,这是Taqman软件中的常见做法。而在我们的这个案例中,我们不会将其替换为40,而是替换为NA值,现在我们读入一个Taqman的qPCR数据,这批数据是用96孔板测的,其中,实验组(mia)4个重复,对照组(no-mia)4个重复,如下所示:
path <- system.file("exData", package = "NormqPCR")
taqman.example <- file.path(path, "/example.txt")
qPCRBatch.taqman <- read.taqman(taqman.example)
现在我们只看某个样本的检测值,即Ccl20.Rn00570287_m1这个数据,如下所示:
exprs(qPCRBatch.taqman)["Ccl20.Rn00570287_m1",]
结果如下所示:
> exprs(qPCRBatch.taqman)["Ccl20.Rn00570287_m1",]
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia fp.3.day.3.v
NA NA 35.74190 34.05922 35.02052
fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
NA 35.93689 36.57921
现在我们使用replaceAboveCutOff()函数将大于35的数值替换为NA,如下所示:
# Use replaceAboveCutOff method in order to replace anything above 35 with NA
qPCRBatch.taqman.replaced <- replaceAboveCutOff(qPCRBatch.taqman,newVal = NA, cutOff = 35)
exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
结果如下所示:
> exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia fp.3.day.3.v
NA NA NA 34.05922 NA
fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
NA NA NA
但是对于有些同学来说,我不想把大于35的数值替换为NA,而是想把所有的NA都替换为40,那么可以使用replaceNAs()函数,如下所示:
# Use replaesNAs methods to replace all NAs with 40
qPCRBatch.taqman.replaced <- replaceNAs(qPCRBatch.taqman, newNA = 40)
exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
结果如下所示:
> exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia fp.3.day.3.v
40.00000 40.00000 35.74190 34.05922 35.02052
fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
40.00000 35.93689 36.57921
有的时候我们会遇到这样的一样种情况,在一次实验中,例如我们做了96孔板的qPCR实验(做了好几块96孔板),某个孔的检测器中检测的数值大于设定的阈值,而有的孔的检测器检测到的数值则没有大于设定的阈值。有的同学可能就会认为,即然一些孔的检测器检测到的值设为NAs,那么这个孔的检测器在读取其它数值时,也应该设为NAs。因为否则一个检测器的计数出现了异学,那么会导致某个基因表达水平的计算出现错误估计(这一段不太理解,原话如下):
In addition, the situation sometimes arises where some readings for a given detector are above a given cycle threshold, but some others are not. The user may decide for example that if a given number of readings are NAs, then all of the readings for this detector should be NAs. This is important because otherwise an unusual reading for one detector might lead to an inaccurate estimate for the expression of a given gene.
对于不同的样本类型这个过程应该分开处理,因为你也许想比较不同样本之间的某个特定的基因。因此有必要使用比较矩阵来指定每个样本类型的重复数。因此,在上面的案例中,我们一共有两个类型,分别是实验组与对照组,每组4个重复,我们可以使用contrastMatrix()函数和sampleMaxMatrix()函数来创建比较矩阵,如下所示:
# Use contrastMatrix() and sampleMaxMatrix() to generate contrast matrix
sampleNames(qPCRBatch.taqman) # Check group information
a <- c(0,0,1,1,0,0,1,1) # one for each sample type, with 1 representing
b <- c(1,1,0,0,1,1,0,0) # position of sample type in samplenames vector
contM <- cbind(a,b)
colnames(contM) <- c("case","control") # set the names of each sample type
rownames(contM) <- sampleNames(qPCRBatch.taqman) # set row names
contM
sMaxM <- t(as.matrix(c(3,3))) # now make the contrast matrix
colnames(sMaxM) <- c("case","control") # make sure these line up with samples
sMaxM
结果如下所示:
> # Use contrastMatrix() and sampleMaxMatrix() to generate contrast matrix
> sampleNames(qPCRBatch.taqman) # Check group information
[1] "fp1.day3.v" "fp2.day3.v" "fp5.day3.mia" "fp6.day3.mia"
[5] "fp.3.day.3.v" "fp.4.day.3.v" "fp.7.day.3.mia" "fp.8.day.3.mia"
> a <- c(0,0,1,1,0,0,1,1) # one for each sample type, with 1 representing
> b <- c(1,1,0,0,1,1,0,0) # position of sample type in samplenames vector
> contM <- cbind(a,b)
> colnames(contM) <- c("case","control") # set the names of each sample type
> rownames(contM) <- sampleNames(qPCRBatch.taqman) # set row names
> contM
case control
fp1.day3.v 0 1
fp2.day3.v 0 1
fp5.day3.mia 1 0
fp6.day3.mia 1 0
fp.3.day.3.v 0 1
fp.4.day.3.v 0 1
fp.7.day.3.mia 1 0
fp.8.day.3.mia 1 0
> sMaxM <- t(as.matrix(c(3,3))) # now make the contrast matrix
> colnames(sMaxM) <- c("case","control") # make sure these line up with samples
> sMaxM
case control
[1,] 3 3
有关更多的算法细节可以参考limmma包中的内容,那个包中创建差异基因比较的矩阵也是类似方法。
例如,如果用户决定将那些4个重复中,有3个是NA的值都设为NA的话,可以使用makeAllNewVal()函数,如下所示:
qPCRBatch.taqman.replaced <- makeAllNewVal(qPCRBatch.taqman, contM,sMaxM, newVal=NA)
现在我们来看一下Ccl20.Rn00570287 m1这个样本的检测值,对照组的值都为NA,因为原始的4个技术重复中,有3个是NA,有一个是35,因此全设为NA。不过实验组的数据全部进行了保留,如下所示:
exprs(qPCRBatch.taqman)["Ccl20.Rn00570287_m1",]
exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
结果如下所示:
> exprs(qPCRBatch.taqman)["Ccl20.Rn00570287_m1",]
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia fp.3.day.3.v
NA NA 35.74190 34.05922 35.02052
fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
NA 35.93689 36.57921
> exprs(qPCRBatch.taqman.replaced)["Ccl20.Rn00570287_m1",]
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia fp.3.day.3.v
NA NA 35.74190 34.05922 NA
fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
NA 35.93689 36.57921
前面这两部分内容(合并技术重复与处理未检测值)了解一下就行,平时也用不到,最重要的是下面的内容。
选择最稳定内参基因
这一部分涉及选择最稳定的内参基因,其中用的是geNorm算法,这个算法的相关文献信息如下所示:
Jo Vandesompele, Katleen De Preter, Filip Pattyn et al. (2002). Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biology 2002. 3(7):research0034.1-0034.11. http://genomebiology.com/2002/3/7/research/0034/
geNorm
先加载数据,如下所示:
options(width = 68)
data(geNorm)
str(exprs(geNorm.qPCRBatch))
数据结构如下所示:
> str(exprs(geNorm.qPCRBatch))
num [1:10, 1:85] 0.0425 0.0576 0.1547 0.1096 0.118 ...
- attr(*, "dimnames")=List of 2
..$ : chr [1:10] "ACTB" "B2M" "GAPD" "HMBS" ...
..$ : chr [1:85] "BM1" "BM2" "BM3" "BM4" ...
现在我们对选择的内参基因进行排序。我们在这里使用selectHKs()函数来实现geNorm算法中的逐步处理过程,这里的数据都是文献中材料与方法中提到的数据。
第一步,我们计算所有内参基因稳定检测值M后,排除那些M值最高的内参基因;
第二步,再次计算M值,再排除掉最高M值的内参基因,直到剩下两个内参基因(也就是参数minNrHK=2,如下所示:
tissue <- as.factor(c(rep("BM", 9), rep("FIB", 20), rep("LEU", 13),rep("NB", 34), rep("POOL", 9)))
res.BM <- selectHKs(geNorm.qPCRBatch[,tissue == "BM"], method = "geNorm",Symbols = featureNames(geNorm.qPCRBatch),minNrHK = 2, log = FALSE)
计算过程中,这个函数会给出不断计算的M值,结果如下所示:
res.BM <- selectHKs(geNorm.qPCRBatch[,tissue == "BM"], method = "geNorm",Symbols = featureNames(geNorm.qPCRBatch),minNrHK = 2, log = FALSE)
###############################################################
Step 1:
stability values M:
HPRT1 YWHAZ RPL13A UBC GAPD SDHA TBP
0.5160313 0.5314564 0.5335963 0.5700961 0.6064919 0.6201470 0.6397969
HMBS B2M ACTB
0.7206013 0.7747634 0.8498739
average stability M: 0.63628545246682
variable with lowest stability (largest M value): ACTB
Pairwise variation, (9/10): 0.0764690052563778
###############################################################
Step 2:
stability values M:
HPRT1 RPL13A YWHAZ UBC GAPD SDHA TBP
0.4705664 0.5141375 0.5271169 0.5554718 0.5575295 0.5738460 0.6042110
HMBS B2M
0.6759176 0.7671985
average stability M: 0.582888329316757
variable with lowest stability (largest M value): B2M
Pairwise variation, (8/9): 0.0776534266912183
###############################################################
Step 3:
stability values M:
HPRT1 RPL13A SDHA YWHAZ UBC GAPD TBP
0.4391222 0.4733732 0.5243665 0.5253471 0.5403137 0.5560120 0.5622094
HMBS
0.6210820
average stability M: 0.530228279613623
variable with lowest stability (largest M value): HMBS
Pairwise variation, (7/8): 0.0671119963410967
###############################################################
Step 4:
stability values M:
HPRT1 RPL13A YWHAZ UBC SDHA GAPD TBP
0.4389069 0.4696398 0.4879728 0.5043292 0.5178634 0.5245346 0.5563591
average stability M: 0.499943693933222
variable with lowest stability (largest M value): TBP
Pairwise variation, (6/7): 0.0681320232188603
###############################################################
Step 5:
stability values M:
HPRT1 RPL13A UBC YWHAZ GAPD SDHA
0.4292808 0.4447874 0.4594181 0.4728920 0.5012107 0.5566762
average stability M: 0.477377523800525
variable with lowest stability (largest M value): SDHA
Pairwise variation, (5/6): 0.0806194432580746
###############################################################
Step 6:
stability values M:
UBC RPL13A HPRT1 YWHAZ GAPD
0.4195958 0.4204997 0.4219179 0.4424631 0.4841646
average stability M: 0.437728198765878
variable with lowest stability (largest M value): GAPD
Pairwise variation, (4/5): 0.0841653121631615
###############################################################
Step 7:
stability values M:
RPL13A UBC YWHAZ HPRT1
0.3699163 0.3978736 0.4173706 0.4419220
average stability M: 0.406770625156432
variable with lowest stability (largest M value): HPRT1
Pairwise variation, (3/4): 0.097678269387021
###############################################################
Step 8:
stability values M:
UBC RPL13A YWHAZ
0.3559286 0.3761358 0.3827933
average stability M: 0.371619241507029
variable with lowest stability (largest M value): YWHAZ
Pairwise variation, (2/3): 0.113745049966055
###############################################################
Step 9:
stability values M:
RPL13A UBC
0.3492712 0.3492712
average stability M: 0.349271187472188
> res.BM
$ranking
1 1 3 4 5 6 7
"RPL13A" "UBC" "YWHAZ" "HPRT1" "GAPD" "SDHA" "TBP"
8 9 10
"HMBS" "B2M" "ACTB"
$variation
9/10 8/9 7/8 6/7 5/6 4/5
0.07646901 0.07765343 0.06711200 0.06813202 0.08061944 0.08416531
3/4 2/3
0.09767827 0.11374505
$meanM
10 9 8 7 6 5
0.6362855 0.5828883 0.5302283 0.4999437 0.4773775 0.4377282
4 3 2
0.4067706 0.3716192 0.3492712
以上只是BM这类组织的计算结果。现在我们把其它的组织都计算出来,如下所示:
res.POOL <- selectHKs(geNorm.qPCRBatch[,tissue == "POOL"],
method = "geNorm",
Symbols = featureNames(geNorm.qPCRBatch),
minNrHK = 2, trace = FALSE, log = FALSE)
res.FIB <- selectHKs(geNorm.qPCRBatch[,tissue == "FIB"],
method = "geNorm",
Symbols = featureNames(geNorm.qPCRBatch),
minNrHK = 2, trace = FALSE, log = FALSE)
res.LEU <- selectHKs(geNorm.qPCRBatch[,tissue == "LEU"],
method = "geNorm",
Symbols = featureNames(geNorm.qPCRBatch),
minNrHK = 2, trace = FALSE, log = FALSE)
res.NB <- selectHKs(geNorm.qPCRBatch[,tissue == "NB"],
method = "geNorm",
Symbols = featureNames(geNorm.qPCRBatch),
minNrHK = 2, trace = FALSE, log = FALSE)
我们就获得了基因的下面排序信息,这个就是文献中的Table 3,文献中的信息如下所示:

我们自己的计算结果:
ranks <- data.frame(c(1, 1:9),
res.BM$ranking, res.POOL$ranking,
res.FIB$ranking, res.LEU$ranking,
res.NB$ranking)
names(ranks) <- c("rank", "BM", "POOL", "FIB", "LEU", "NB")
ranks
结果如下所示:
ranks
rank BM POOL FIB LEU NB
1 1 RPL13A GAPD GAPD UBC GAPD
2 1 UBC SDHA HPRT1 YWHAZ HPRT1
3 2 YWHAZ HMBS YWHAZ B2M SDHA
4 3 HPRT1 HPRT1 UBC GAPD UBC
5 4 GAPD TBP ACTB RPL13A HMBS
6 5 SDHA UBC TBP TBP YWHAZ
7 6 TBP RPL13A SDHA SDHA TBP
8 7 HMBS YWHAZ RPL13A HPRT1 ACTB
9 8 B2M ACTB B2M HMBS RPL13A
10 9 ACTB B2M HMBS ACTB B2M
现在绘制出每个细胞类型的平均基因表达稳定值M的折线图,这个对应于文献中的Figure 2,如下所示:

代码如下所示:
library(RColorBrewer)
mypalette <- brewer.pal(5, "Set1")
matplot(cbind(res.BM$meanM, res.POOL$meanM, res.FIB$meanM,
res.LEU$meanM, res.NB$meanM), type = "b",
ylab = "Average expression stability M",
xlab = "Number of remaining control genes",
axes = FALSE, pch = 19, col = mypalette,
ylim = c(0.2, 1.22), lty = 1, lwd = 2,
main = "Figure 2 in Vandesompele et al. (2002)")
axis(1, at = 1:9, labels = as.character(10:2))
axis(2, at = seq(0.2, 1.2, by = 0.2), labels = seq(0.2, 1.2, by = 0.2))
box()
abline(h = seq(0.2, 1.2, by = 0.2), lty = 2, lwd = 1, col = "grey")
legend("topright", legend = c("BM", "POOL", "FIB", "LEU", "NB"),
fill = mypalette)

绘制每个细胞类型的成对方差(pairwise variation)图,这个对应文献中的Figure 3,如下所示:
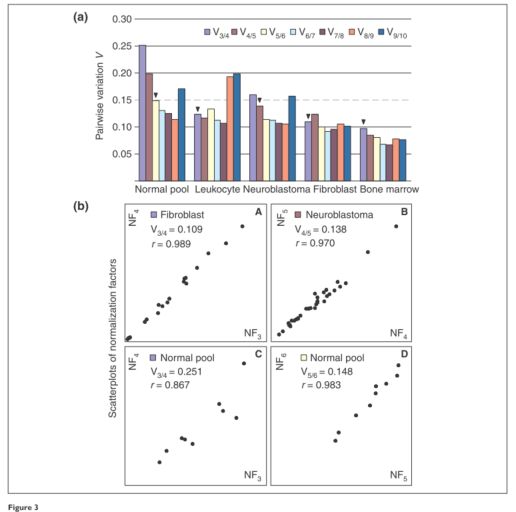
代码如下所示:
mypalette <- brewer.pal(8, "YlGnBu")
barplot(cbind(res.POOL$variation, res.LEU$variation, res.NB$variation,
res.FIB$variation, res.BM$variation), beside = TRUE,
col = mypalette, space = c(0, 2),
names.arg = c("POOL", "LEU", "NB", "FIB", "BM"),
ylab = "Pairwise variation V",
main = "Figure 3(a) in Vandesompele et al. (2002)")
legend("topright", legend = c("V9/10", "V8/9", "V7/8", "V6/7",
"V5/6", "V4/5", "V3/4", "V2/3"),
fill = mypalette, ncol = 2)
abline(h = seq(0.05, 0.25, by = 0.05), lty = 2, col = "grey")
abline(h = 0.15, lty = 1, col = "black")
注:文献中推荐的成对方差值的截止值为0.15,因此低于此值后,就不需要使用其它的内参基因了。

NormFinder
第二种选择内参的基因的方法是NormFinder,这种方法来源于以下文献:
Andersen, C. L., et al. (2004). "Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets." Cancer Res 64(15): 5245-5250.
现在我们计算Table 3,如下所示:

第一步,导入数据::
## NormFinder
data(Colon)
Colon
结果如下所示:
> data(Colon)
> Colon
qPCRBatch (storageMode: lockedEnvironment)
assayData: 13 features, 40 samples
element names: exprs
protocolData: none
phenoData
sampleNames: I459N 90 ... I-C1056T (40 total)
varLabels: Sample.no. Classification
varMetadata: labelDescription
featureData
featureNames: UBC UBB ... TUBA6 (13 total)
fvarLabels: Symbol Gene.name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: Table 1 in Andersen et al. (2004)
继续计算,如下所示:
Class <- pData(Colon)[,"Classification"]
res.Colon <- stabMeasureRho(Colon, group = Class, log = FALSE)
sort(res.Colon) # see Table 3 in Andersen et al (2004)
结果如下所示:
> sort(res.Colon) # see Table 3 in Andersen et al (2004)
UBC GAPD TPT1 UBB TUBA6 RPS13 NACA
0.1821707 0.2146061 0.2202956 0.2471573 0.2700641 0.2813039 0.2862397
CFL1 SUI1 ACTB CLTC RPS23 FLJ20030
0.2870467 0.3139404 0.3235918 0.3692880 0.3784909 0.3935173
再看另外的一个数据集Bladder,如下所示:
data(Bladder)
Bladder
grade <- pData(Bladder)[,"Grade"]
res.Bladder <- stabMeasureRho(Bladder,
group = grade,
log = FALSE)
sort(res.Bladder)
计算结果如下所示:
> data(Bladder)
> Bladder
qPCRBatch (storageMode: lockedEnvironment)
assayData: 14 features, 28 samples
element names: exprs
protocolData: none
phenoData
sampleNames: 335-6 1131-1 ... 1356-1 (28 total)
varLabels: Sample.no. Grade
varMetadata: labelDescription
featureData
featureNames: ATP5B HSPCB ... FLJ20030 (14 total)
fvarLabels: Symbol Gene.name
fvarMetadata: labelDescription
experimentData: use 'experimentData(object)'
Annotation: Table 1 in Andersen et al. (2004)
> grade <- pData(Bladder)[,"Grade"]
> res.Bladder <- stabMeasureRho(Bladder,
+ group = grade,
+ log = FALSE)
Warning message:
In .local(x, ...) : Argument 'group' is transformed to a factor vector.
> sort(res.Bladder)
HSPCB TEGT ATP5B UBC RPS23 RPS13 CFL1
0.1539598 0.1966556 0.1987227 0.2033477 0.2139626 0.2147852 0.2666129
FLJ20030 TPT1 UBB FLOT2 GAPD S100A6 ACTB
0.2672918 0.2691553 0.2826051 0.2960429 0.3408742 0.3453435 0.3497295
我们也可以使用geNorm算法来计算,如下所示:
selectHKs(Colon,
log = FALSE,
trace = FALSE,
Symbols = featureNames(Colon))$ranking
计算结果如下所示:
> selectHKs(Colon,
+ log = FALSE,
+ trace = FALSE,
+ Symbols = featureNames(Colon))$ranking
1 1 3 4 5 6 7
"RPS23" "TPT1" "RPS13" "SUI1" "UBC" "GAPD" "TUBA6"
8 9 10 11 12 13
"UBB" "NACA" "CFL1" "CLTC" "ACTB" "FLJ20030"
再来看Baldder的数据,如下所示:
selectHKs(Bladder,
log = FALSE,
trace = FALSE,
Symbols = featureNames(Bladder))$ranking
结果如下所示:
> selectHKs(Bladder,
+ log = FALSE,
+ trace = FALSE,
+ Symbols = featureNames(Bladder))$ranking
1 1 3 4 5 6 7
"CFL1" "UBC" "ATP5B" "HSPCB" "GAPD" "TEGT" "RPS23"
8 9 10 11 12 13 14
"RPS13" "TPT1" "FLJ20030" "FLOT2" "UBB" "ACTB" "S100A6"
我们也可以使用NormFinder算法的分步计算方式工来看多个内参基因的情况,也就是文献中附录中的Average control gene部分,selectHKs函数中含有这种算法的参数,如下所示:
Class <- pData(Colon)[,"Classification"]
selectHKs(Colon,
group = Class,
log = FALSE,
trace = TRUE,
Symbols = featureNames(Colon),
minNrHKs = 12,
method = "NormFinder")$ranking
结果如下所示:
> Class <- pData(Colon)[,"Classification"]
> selectHKs(Colon,
+ group = Class,
+ log = FALSE,
+ trace = TRUE,
+ Symbols = featureNames(Colon),
+ minNrHKs = 12,
+ method = "NormFinder")$ranking
###############################################################
Step 1:
stability values rho:
UBC GAPD TPT1 UBB TUBA6 RPS13 NACA
0.1821707 0.2146061 0.2202956 0.2471573 0.2700641 0.2813039 0.2862397
CFL1 SUI1 ACTB CLTC RPS23 FLJ20030
0.2870467 0.3139404 0.3235918 0.3692880 0.3784909 0.3935173
variable with highest stability (smallest rho value): UBC
###############################################################
Step 2:
stability values rho:
GAPD TPT1 UBB NACA CFL1 RPS13 TUBA6
0.1375298 0.1424519 0.1578360 0.1657364 0.1729069 0.1837057 0.1849021
SUI1 ACTB RPS23 FLJ20030 CLTC
0.2065531 0.2131651 0.2188277 0.2359623 0.2447588
variable with highest stability (smallest rho value): GAPD
###############################################################
Step 3:
stability values rho:
TPT1 NACA UBB RPS13 CFL1 TUBA6 FLJ20030
0.1108474 0.1299802 0.1356690 0.1411173 0.1474242 0.1532953 0.1583031
SUI1 ACTB RPS23 CLTC
0.1586250 0.1682972 0.1686139 0.1926907
variable with highest stability (smallest rho value): TPT1
###############################################################
Step 4:
stability values rho:
UBB TUBA6 ACTB CFL1 RPS13 SUI1 CLTC
0.09656546 0.09674897 0.10753445 0.10830099 0.11801680 0.12612399 0.12773131
NACA FLJ20030 RPS23
0.13422958 0.14609897 0.16530522
variable with highest stability (smallest rho value): UBB
###############################################################
Step 5:
stability values rho:
RPS13 SUI1 TUBA6 NACA FLJ20030 CFL1 ACTB
0.09085973 0.09647829 0.09943424 0.10288912 0.11097074 0.11428399 0.11495336
RPS23 CLTC
0.12635109 0.13286210
variable with highest stability (smallest rho value): RPS13
###############################################################
Step 6:
stability values rho:
ACTB TUBA6 CFL1 FLJ20030 NACA CLTC SUI1
0.09215478 0.09499893 0.09674032 0.10528784 0.10718604 0.10879846 0.11368091
RPS23
0.13134766
variable with highest stability (smallest rho value): ACTB
###############################################################
Step 7:
stability values rho:
SUI1 NACA FLJ20030 RPS23 TUBA6 CFL1 CLTC
0.08281504 0.08444905 0.08922236 0.09072667 0.10559279 0.10993755 0.13142181
variable with highest stability (smallest rho value): SUI1
###############################################################
Step 8:
stability values rho:
NACA CFL1 TUBA6 FLJ20030 CLTC RPS23
0.08336046 0.08410148 0.09315528 0.09775742 0.10499056 0.10554332
variable with highest stability (smallest rho value): NACA
###############################################################
Step 9:
stability values rho:
CFL1 TUBA6 CLTC FLJ20030 RPS23
0.07222968 0.07722737 0.08440691 0.09831958 0.12735605
variable with highest stability (smallest rho value): CFL1
###############################################################
Step 10:
stability values rho:
FLJ20030 TUBA6 CLTC RPS23
0.08162006 0.08189011 0.10705192 0.11430674
variable with highest stability (smallest rho value): FLJ20030
###############################################################
Step 11:
stability values rho:
TUBA6 CLTC RPS23
0.06978897 0.08069582 0.13702726
variable with highest stability (smallest rho value): TUBA6
###############################################################
Step 12:
stability values rho:
CLTC RPS23
0.1199009 0.1245241
variable with highest stability (smallest rho value): CLTC
1 2 3 4 5 6 7
"UBC" "GAPD" "TPT1" "UBB" "RPS13" "ACTB" "SUI1"
8 9 10 11 12
"NACA" "CFL1" "FLJ20030" "TUBA6" "CLTC"
Warning message:
In .local(x, ...) : Argument 'group' is transformed to a factor vector.
在Bladder数据集中,我们发现,排列前面的2个内参基因是HSPCB与RPS13,也就是Figure 1中的内容,如下所示:

grade <- pData(Bladder)[,"Grade"]
selectHKs(Bladder,
group = grade,
log = FALSE,
trace = FALSE,
Symbols = featureNames(Bladder),
minNrHKs = 13,
method = "NormFinder")$ranking
结果如下所示:
> grade <- pData(Bladder)[,"Grade"]
> selectHKs(Bladder,
+ group = grade,
+ log = FALSE,
+ trace = FALSE,
+ Symbols = featureNames(Bladder),
+ minNrHKs = 13,
+ method = "NormFinder")$ranking
1 2 3 4 5 6 7
"HSPCB" "RPS13" "UBC" "RPS23" "ATP5B" "TEGT" "UBB"
8 9 10 11 12 13
"FLJ20030" "CFL1" "S100A6" "FLOT2" "ACTB" "TPT1"
使用多内参进行均一化
单内参的ΔCq方法
单内参均一化的方法就是,使用某个基因的Cq值减去内参的Cq值。NormqPCR中就有相关的计算函数,如下所示:
path <- system.file("exData", package = "NormqPCR")
taqman.example <- file.path(path, "example.txt")
qPCR.example <- file.path(path, "qPCR.example.txt")
qPCRBatch.taqman <- read.taqman(taqman.example)
hkgs<-"Actb-Rn00667869_m1"
qPCRBatch.norm <- deltaCq(qPCRBatch = qPCRBatch.taqman, hkgs = hkgs, calc="arith")
head(exprs(qPCRBatch.taqman)) # raw data
head(exprs(qPCRBatch.norm)) # Normlazation data
这里我们使用了Actb作为内参,计算结果如下所示:
> head(exprs(qPCRBatch.taqman)) # raw data
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia
Actb.Rn00667869_m1 20.92301 21.00262 20.88438 20.38190
Adipoq.Rn00595250_m1 20.93906 20.88610 23.81790 22.92289
Adrbk1.Rn00562822_m1 NA NA 27.45101 27.02446
Agtrl1.Rn00580252_s1 25.82239 26.03846 27.28175 26.06274
Alpl.Rn00564931_m1 33.45495 32.81128 33.91955 32.62145
B2m.Rn00560865_m1 21.66457 21.89334 22.92485 22.61651
fp.3.day.3.v fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
Actb.Rn00667869_m1 20.94854 21.48838 20.97863 21.10281
Adipoq.Rn00595250_m1 20.76957 20.92512 23.43714 23.83928
Adrbk1.Rn00562822_m1 NA NA 24.71573 27.97638
Agtrl1.Rn00580252_s1 26.16934 25.91374 25.77341 26.44801
Alpl.Rn00564931_m1 33.34335 33.26128 33.08863 33.35799
B2m.Rn00560865_m1 21.45406 22.36598 22.90620 23.00608
> head(exprs(qPCRBatch.norm)) # Normlazation data
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia
Actb.Rn00667869_m1 0.000000 0.000000 0.000000 0.000000
Adipoq.Rn00595250_m1 0.016052 -0.116520 2.933523 2.540987
Adrbk1.Rn00562822_m1 NA NA 6.566628 6.642561
Agtrl1.Rn00580252_s1 4.899380 5.035841 6.397364 5.680837
Alpl.Rn00564931_m1 12.531942 11.808657 13.035166 12.239549
B2m.Rn00560865_m1 0.741558 0.890717 2.040470 2.234605
fp.3.day.3.v fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
Actb.Rn00667869_m1 0.000000 0.000000 0.000000 0.000000
Adipoq.Rn00595250_m1 -0.178971 -0.563263 2.458509 2.736475
Adrbk1.Rn00562822_m1 NA NA 3.737100 6.873568
Agtrl1.Rn00580252_s1 5.220796 4.425364 4.794776 5.345202
Alpl.Rn00564931_m1 12.394802 11.772896 12.110000 12.255186
B2m.Rn00560865_m1 0.505516 0.877598 1.927563 1.903269
生成的结果是一个qPCRBatch对象,很多R包例如heatmap就兼容这类对象。
多内参的ΔCq方法
如果使用前面提到的geNorm或NormFinder算法找到了多个内参,此时想要用这多个内参进行均一化,那么就需要计算这几个内参的均值,构成一个“伪内参“(pseudo-housekeeper)进行均一化。现在我们使用三个内参,分别为GAPDH,beta-2-microglobulin和Beta-actin来进行均一化,如下所示:
hkgs<-c("Actb-Rn00667869_m1", "B2m-Rn00560865_m1", "Gapdh-Rn99999916_s1")
qPCRBatch.norm <- deltaCq(qPCRBatch = qPCRBatch.taqman, hkgs = hkgs, calc="arith")
head(exprs(qPCRBatch.norm))
结果如下所示:
> head(exprs(qPCRBatch.norm))
fp1.day3.v fp2.day3.v fp5.day3.mia fp6.day3.mia
Actb.Rn00667869_m1 -1.2998917 -1.2816963 -1.380296 -1.5106197
Adipoq.Rn00595250_m1 -1.2838397 -1.3982163 1.553227 1.0303673
Adrbk1.Rn00562822_m1 NA NA 5.186332 5.1319413
Agtrl1.Rn00580252_s1 3.5994883 3.7541447 5.017068 4.1702173
Alpl.Rn00564931_m1 11.2320503 10.5269607 11.654870 10.7289293
B2m.Rn00560865_m1 -0.5583337 -0.3909793 0.660174 0.7239853
fp.3.day.3.v fp.4.day.3.v fp.7.day.3.mia fp.8.day.3.mia
Actb.Rn00667869_m1 -1.1644617 -1.1714227 -1.323712 -1.286277
Adipoq.Rn00595250_m1 -1.3434327 -1.7346857 1.134797 1.450198
Adrbk1.Rn00562822_m1 NA NA 2.413388 5.587291
Agtrl1.Rn00580252_s1 4.0563343 3.2539413 3.471064 4.058925
Alpl.Rn00564931_m1 11.2303403 10.6014733 10.786288 10.968909
B2m.Rn00560865_m1 -0.6589457 -0.2938247 0.603851 0.616992
单内参2^-ΔΔCq方法
使用2^-ΔΔCq方法哦可以计算两个样本类型之间的相对表达值。默认情况下,同一个样本的目标基因与其内参基因配对进行计算,此时的标准差就体现在不同基因与不同内参上。但是,如果参数中设定了paired=FALSE,那么标准差的计算公式就是s=sqrt(s1^2+s2^2),这里的s1是目标基因的标准差,s2是内参基因的标准差,但是,如果当你要比较的基因与内参基因是来源于同一个样本时,不推荐这种方法,例如使用Taqman板的情况就是如此,但是,如果要考虑到完整性(completeness)以及要使用不同的生物学重复中获得的内参基因时,可以考虑使用这种计算方法,例如我们最初设计的内参数据并不好时,当采用事后检验(post hoc)时,就采用这种方法,或者是当NormqPCR用于处理那些单独板孔中的扩增数据时,也是采用这种方法。
现在我们读取原始数据,如下所示:
path <- system.file("exData", package = "NormqPCR")
taqman.example <- file.path(path, "example.txt")
qPCR.example <- file.path(path, "qPCR.example.txt")
qPCRBatch.taqman <- read.taqman(taqman.example)
deltaDeltaCq()函数在进行计算时需要一个比较矩阵。在这个矩阵中,列是样本名(例如case或control),这与limma包中的数据类似。这个比较矩阵的列中只包括1或0这两个数据,用于指定分类变量,如下所示:
contM <- cbind(c(0,0,1,1,0,0,1,1),c(1,1,0,0,1,1,0,0))
colnames(contM) <- c("interestingPhenotype","wildTypePhenotype")
rownames(contM) <- sampleNames(qPCRBatch.taqman)
contM
结果如下所示:
> contM
interestingPhenotype wildTypePhenotype
fp1.day3.v 0 1
fp2.day3.v 0 1
fp5.day3.mia 1 0
fp6.day3.mia 1 0
fp.3.day.3.v 0 1
fp.4.day.3.v 0 1
fp.7.day.3.mia 1 0
fp.8.day.3.mia 1 0
此时,我们可以指定一个内参基因来进行均一化,然后再看一个某个基因在实验组(case)和对照组(control)的比值,计算结果包括以下信息(按列显示):
- 每个基因的表达值;
- 实验组的ΔCq值:
- Cq值的sd;
- 对照组的ΔCq值;
- 对照组的ΔCq值的sd;
- 2^-ΔΔCq值,这是对照组与实验组的差异;
- 和8. 分别是1和0时的sd值,如下所示:
hkg <- "Actb-Rn00667869_m1"
ddCq.taqman <- deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
maxNACase=1, maxNAControl=1,
hkg=hkg,
contrastM=contM,
case="interestingPhenotype",
control="wildTypePhenotype",
statCalc="geom",
hkgCalc="arith")
head(ddCq.taqman)
结果如下所示:
> head(ddCq.taqman)
ID 2^-dCt.interestingPhenotype interestingPhenotype.sd
1 Actb.Rn00667869_m1 1.000e+00 0.000e+00
2 Adipoq.Rn00595250_m1 1.587e-01 2.280e-02
3 Adrbk1.Rn00562822_m1 2.602e-02 3.266e-02
4 Agtrl1.Rn00580252_s1 2.300e-02 1.014e-02
5 Alpl.Rn00564931_m1 1.892e-04 4.770e-05
6 B2m.Rn00560865_m1 2.464e-01 2.498e-02
2^-dCt.wildTypePhenotype wildTypePhenotype.sd 2^-ddCt 2^-ddCt.min
1 1.000e+00 0.000e+00 1 NA
2 1.171e+00 2.131e-01 0.135541545192243 NA
3 NA NA + NA
4 3.434e-02 8.584e-03 0.669721905042939 NA
5 2.298e-04 6.107e-05 0.823327272466571 NA
6 5.965e-01 7.668e-02 0.413128242070071 NA
2^-ddCt.max
1 NA
2 NA
3 NA
4 NA
5 NA
6 NA
也可以使用每个单独的样本/孔(samples/wells)方法来计算平均值。这里我们单独计算sd,然后将它们汇总起来。因此,同一个样本的内参与检测值的本领信息氷是人干活的,这同时会增加变异,如下所示:
hkg <- "Actb-Rn00667869_m1"
ddCqAvg.taqman <- deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
maxNACase=1,
maxNAControl=1,
hkg=hkg,
contrastM=contM,
case="interestingPhenotype",
control="wildTypePhenotype",
paired=FALSE, statCalc="geom",
hkgCalc="arith")
head(ddCqAvg.taqman)
运行结果如下所示:
> hkg <- "Actb-Rn00667869_m1"
> ddCqAvg.taqman <- deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
+ maxNACase=1,
+ maxNAControl=1,
+ hkg=hkg,
+ contrastM=contM,
+ case="interestingPhenotype",
+ control="wildTypePhenotype",
+ paired=FALSE, statCalc="geom",
+ hkgCalc="arith")
There were 12 warnings (use warnings() to see them)
>
> head(ddCqAvg.taqman)
ID 2^-dCt.interestingPhenotype interestingPhenotype.sd
1 Actb.Rn00667869_m1 1.000e+00 0.000e+00
2 Adipoq.Rn00595250_m1 1.587e-01 2.280e-02
3 Adrbk1.Rn00562822_m1 2.602e-02 3.266e-02
4 Agtrl1.Rn00580252_s1 2.300e-02 1.014e-02
5 Alpl.Rn00564931_m1 1.892e-04 4.770e-05
6 B2m.Rn00560865_m1 2.464e-01 2.498e-02
2^-dCt.wildTypePhenotype wildTypePhenotype.sd 2^-ddCt 2^-ddCt.min
1 1.000e+00 0.000e+00 1 NA
2 1.171e+00 2.131e-01 0.135541545192243 NA
3 NA NA + NA
4 3.434e-02 8.584e-03 0.669721905042939 NA
5 2.298e-04 6.107e-05 0.823327272466571 NA
6 5.965e-01 7.668e-02 0.413128242070071 NA
2^-ddCt.max
1 NA
2 NA
3 NA
4 NA
5 NA
6 NA
多内参2^-ΔΔCq方法
如果想使用大于1个内参进行均一化,例如,我们想使用NormFinder/geNorm算法来进行均一化。我们就可以使用几何平均数(geometric mean)来构建一个伪内参(pseudo-housekeeper)来实现这一目的。现在我们使用GAPDH,beta-2-microglobulin和Beta-actin这三个内参来进行均一化,如下所示:
qPCRBatch.taqman <- read.taqman(taqman.example)
contM <- cbind(c(0,0,1,1,0,0,1,1),c(1,1,0,0,1,1,0,0))
colnames(contM) <- c("interestingPhenotype","wildTypePhenotype")
rownames(contM) <- sampleNames(qPCRBatch.taqman)
hkgs<-c("Actb-Rn00667869_m1", "B2m-Rn00560865_m1", "Gapdh-Rn99999916_s1")
ddCq.gM.taqman <- deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
maxNACase=1,
maxNAControl=1,
hkgs=hkgs,
contrastM=contM,
case="interestingPhenotype",
control="wildTypePhenotype",
statCalc="arith",
hkgCalc="arith")
head(ddCq.gM.taqman)
计算结果如下所示:
> qPCRBatch.taqman <- read.taqman(taqman.example)
Warning message:
In read.taqman(taqman.example) :
Incompatible phenoData object. Created a new one using sample name data derived from raw data.
> contM <- cbind(c(0,0,1,1,0,0,1,1),c(1,1,0,0,1,1,0,0))
> colnames(contM) <- c("interestingPhenotype","wildTypePhenotype")
> rownames(contM) <- sampleNames(qPCRBatch.taqman)
> hkgs<-c("Actb-Rn00667869_m1", "B2m-Rn00560865_m1", "Gapdh-Rn99999916_s1")
> ddCq.gM.taqman <- deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
+ maxNACase=1,
+ maxNAControl=1,
+ hkgs=hkgs,
+ contrastM=contM,
+ case="interestingPhenotype",
+ control="wildTypePhenotype",
+ statCalc="arith",
+ hkgCalc="arith")
There were 12 warnings (use warnings() to see them)
> head(ddCq.gM.taqman)
ID 2^-dCt.interestingPhenotype interestingPhenotype.sd
1 Actb.Rn00667869_m1 2.594e+00 0.09819
2 Adipoq.Rn00595250_m1 4.083e-01 0.24929
3 Adrbk1.Rn00562822_m1 4.182e-02 1.45844
4 Agtrl1.Rn00580252_s1 5.520e-02 0.63719
5 Alpl.Rn00564931_m1 4.767e-04 0.42589
6 B2m.Rn00560865_m1 6.367e-01 0.05413
2^-dCt.wildTypePhenotype wildTypePhenotype.sd 2^-ddCt 2^-ddCt.min
1 2.345e+00 0.071373 1.10638851325547 1.034e+00
2 2.713e+00 0.201905 0.150497255530234 1.266e-01
3 NA NA + NA
4 7.878e-02 0.333840 0.700597907024805 4.505e-01
5 5.242e-04 0.386280 0.909381199520663 6.769e-01
6 1.390e+00 0.163975 0.457939394245865 4.411e-01
2^-ddCt.max
1 1.184310
2 0.178884
3 NA
4 1.089636
5 1.221662
6 0.475448
我们也可以使用那些在样本间方差相同的内参来进行均一化,这个类似于前面提到的第二个deltaDeltaCq方法,如下所示:
qPCRBatch.taqman <- read.taqman(taqman.example)
contM <- cbind(c(0,0,1,1,0,0,1,1),c(1,1,0,0,1,1,0,0))
colnames(contM) <- c("interestingPhenotype","wildTypePhenotype")
rownames(contM) <- sampleNames(qPCRBatch.taqman)
hkgs<-c("Actb-Rn00667869_m1", "B2m-Rn00560865_m1", "Gapdh-Rn99999916_s1")
ddAvgCq.gM.taqman <-deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
maxNACase=1, maxNAControl=1,
hkgs=hkgs,
contrastM=contM,
case="interestingPhenotype",
control="wildTypePhenotype",
paired=FALSE,
statCalc="arith",
hkgCalc="arith")
head(ddAvgCq.gM.taqman)
计算结果如下所示:
> qPCRBatch.taqman <- read.taqman(taqman.example)
Warning message:
In read.taqman(taqman.example) :
Incompatible phenoData object. Created a new one using sample name data derived from raw data.
> contM <- cbind(c(0,0,1,1,0,0,1,1),c(1,1,0,0,1,1,0,0))
> colnames(contM) <- c("interestingPhenotype","wildTypePhenotype")
> rownames(contM) <- sampleNames(qPCRBatch.taqman)
> hkgs<-c("Actb-Rn00667869_m1", "B2m-Rn00560865_m1", "Gapdh-Rn99999916_s1")
> ddAvgCq.gM.taqman <-deltaDeltaCq(qPCRBatch = qPCRBatch.taqman,
+ maxNACase=1, maxNAControl=1,
+ hkgs=hkgs,
+ contrastM=contM,
+ case="interestingPhenotype",
+ control="wildTypePhenotype",
+ paired=FALSE,
+ statCalc="arith",
+ hkgCalc="arith")
There were 12 warnings (use warnings() to see them)
> head(ddAvgCq.gM.taqman)
ID 2^-dCt.interestingPhenotype interestingPhenotype.sd
1 Actb.Rn00667869_m1 2.594e+00 0.3849
2 Adipoq.Rn00595250_m1 4.083e-01 0.4822
3 Adrbk1.Rn00562822_m1 4.182e-02 1.4545
4 Agtrl1.Rn00580252_s1 5.520e-02 0.6905
5 Alpl.Rn00564931_m1 4.767e-04 0.5846
6 B2m.Rn00560865_m1 6.367e-01 0.2777
2^-dCt.wildTypePhenotype wildTypePhenotype.sd 2^-ddCt 2^-ddCt.min
1 2.345e+00 0.3574 1.10638851325547 8.473e-01
2 2.713e+00 0.2495 0.150497255530234 1.077e-01
3 NA NA + NA
4 7.878e-02 0.2813 0.700597907024805 4.341e-01
5 5.242e-04 0.3689 0.909381199520663 6.064e-01
6 1.390e+00 0.4576 0.457939394245865 3.778e-01
2^-ddCt.max
1 1.444684
2 0.210221
3 NA
4 1.130625
5 1.363762
6 0.555126
NRQ值计算
导入数据,如下所示:
path <- system.file("exData", package = "ReadqPCR")
qPCR.example <- file.path(path, "qPCR.example.txt")
Cq.data <- read.qPCR(qPCR.example)
合并技术重复,并计算SD,如下所示:
Cq.data1 <- combineTechRepsWithSD(Cq.data)
加载数据集的效应(efficiencies),并将它们添加到数据集中,如下所示:
Effs <- file.path(path, "Efficiencies.txt")
Cq.effs <- read.table(file = Effs, row.names = 1, header = TRUE)
rownames(Cq.effs) <- featureNames(Cq.data1)
effs(Cq.data1) <- as.matrix(Cq.effs[,"efficiency",drop = FALSE])
se.effs(Cq.data1) <- as.matrix(Cq.effs[,"SD.efficiency",drop = FALSE])
计算数据集的NRQ(normalized relative quantities),我们只考虑两个特征值作为内参基因,如下所示:
res <- ComputeNRQs(Cq.data1, hkgs = c("gene_az", "gene_gx"))
## NRQs
exprs(res)
## SD of NRQs
se.exprs(res)
结果如下所示:
> res <- ComputeNRQs(Cq.data1, hkgs = c("gene_az", "gene_gx"))
Warning message:
In .local(qPCRBatch, ...) : This function is still experimental!
> ## NRQs
> exprs(res)
caseA caseB controlA controlB
gene_ai 1.9253072 1.3586729 0.6479659 0.8749479
gene_az 1.0567118 1.1438982 1.0331980 0.9134997
gene_bc 1.1024935 0.7193500 0.7030487 1.2140836
gene_by 1.5102316 0.9573047 0.7527082 1.6008850
gene_dh 1.2982037 1.0722522 0.9623335 0.9392871
gene_dm 0.6590246 1.1690720 1.2475372 0.9366210
gene_dq 0.7541955 0.7036408 0.8327917 1.6165326
gene_dr 2.2192305 1.0581211 0.7026411 0.6900584
gene_eg 0.9366671 0.5800339 0.8313720 1.1848856
gene_er 0.5269062 0.9375427 0.6953326 2.3195978
gene_ev 1.4622280 2.3457021 0.9038912 1.1454535
gene_fr 1.4954763 1.6200792 0.9641192 0.7295680
gene_fw 0.6944248 0.8051075 1.5698382 0.7978611
gene_gx 0.9463318 0.8742037 0.9678687 1.0946911
gene_hl 1.0009372 1.4015267 0.7683665 0.7713712
gene_il 1.4632019 1.2595559 0.7216891 0.9318860
gene_iv 1.7263335 1.2275001 1.5464212 0.8881605
gene_jr 0.8984351 0.9834026 0.8754813 0.6637941
gene_jw 1.4655948 0.9340184 1.0505200 1.5504136
gene_qs 0.6730225 0.7610418 1.0665938 3.5329891
gene_qy 0.5287127 1.5722670 1.0615326 3.3252907
gene_rz 0.8690600 1.5588299 0.7287288 1.4812753
gene_sw 0.5975288 1.2406438 0.6982954 1.6007333
gene_vx 0.6942254 0.7168408 2.0253177 1.3190943
gene_xz 0.7668030 1.0218209 0.6136038 1.6729352
> ## SD of NRQs
> se.exprs(res)
caseA caseB controlA controlB
gene_ai 1.3996554 0.8787290 0.4855882 1.0034912
gene_az 0.6832730 0.7601966 0.8971054 0.6031927
gene_bc 0.7225348 0.4746146 0.9626570 1.0478385
gene_by 1.1522746 0.6116269 0.6088836 2.0409211
gene_dh 1.2483072 0.7889984 0.6165041 0.9767947
gene_dm 0.4711409 0.7780238 0.8476053 0.7294405
gene_dq 0.7023561 0.4849899 0.5813310 1.4067670
gene_dr 1.4407662 1.0804211 0.4543153 0.5149367
gene_eg 0.7355269 0.5497433 0.5588801 1.0938601
gene_er 0.4301195 0.6119514 0.4471454 1.5115897
gene_ev 1.0094209 2.4267114 0.6337126 0.7782519
gene_fr 1.6760391 1.1119157 0.6226081 0.5040967
gene_fw 0.5041070 0.9131565 1.1153268 0.7234551
gene_gx 0.6046042 0.9027816 0.6713914 1.7394961
gene_hl 0.7633174 0.9123997 1.0000329 0.5005813
gene_il 1.4621406 0.9540445 0.5678634 0.6067147
gene_iv 1.2668346 0.8039841 0.9995225 0.8171996
gene_jr 0.5749672 0.6786989 0.5595295 0.4405919
gene_jw 0.9626606 0.7890401 0.7194378 1.1512512
gene_qs 0.5830335 0.5309990 0.6828952 2.8253799
gene_qy 0.5947918 1.1294199 0.6794829 2.1713340
gene_rz 0.5846751 1.7926435 0.4911506 2.1424580
gene_sw 0.6284440 0.8062083 0.9638307 1.8398593
gene_vx 0.5285361 1.0126959 1.3861226 0.8683886
gene_xz 0.5231477 0.9270275 0.3972901 1.3643840
参考资料
- NormqPCR: Functions for normalisation of RT-qPCR data