DAS工具: 利用去重、聚合和评分的策略从宏基因组中恢复基因组
推荐指数:★★★★★
阅读时间:6 分钟
文本字数:2260字
推荐理由:
佳作推荐
文章介绍了一种去重、聚合和评分策略——DAS工具,它灵活的结合了已建立的binning算法的优点。将DAS工具应用于构建的群落可以生成比任何自动化方法更精确的bins。实际上,当应用于不同复杂程度的环境和宿主相关样本时,DAS工具能够恢复更多接近完整的基因组,优于使用任何单一的binning方法,且包括以前未报道的谱系。
利用去重、聚合和评分的策略从宏基因组中恢复基因组
Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy
作者:Christian M. K. Sieber, Alexander J. Probst, et al.
期刊:Nature Microbiology
时间:2018.5
IF: 14.174
DOI: 10.1038/s41564-018-0171-1
研究背景
微生物群落对生态系统功能至关重要。宏基因组学研究的一个关键目标是分析物种特异性的代谢途径,并重建群落代谢互作网络。目前,从宏基因组中重构单菌基因组是实现这一目标的重要手段和方法,这需要将组装好的宏基因组片段精确地分配给每个基因组(binning),从而推断各个物种的代谢途径,并预测其在微生物群落中的生活方式。现有的binning方法往往不能重建合理数量的基因组,并且会产生许多质量和完整性差的基因组。此外,没有一种方法在所有生态系统中表现良好。总之,不同的binning工具具有不同性能以及各自重建具有不同完整度水平的基因组。由此,本研究开发了一套整合多个binning算法的预测结果的策略——DAS工具。
研究方法
依次使用5种常用binning工具(ABAWACA、CONCOCT、Maxbin,、MetaBAT、ESOM),以及作者开发的将5种binning结果结合的DAS工具应用于不同微生物群落:
1. 模拟微生物群落
创建三种不同复杂程度的微生物群落,分别包括低(40个基因组),中(132个基因组)和高复杂度(596个基因组)的微生物群落。
2. 环境宏基因组
选择成人肠道、原油渗漏和土壤环境的宏基因组分别代表较低、中、高复杂度微生物群落。
研究结果
1. DAS工具算法总览
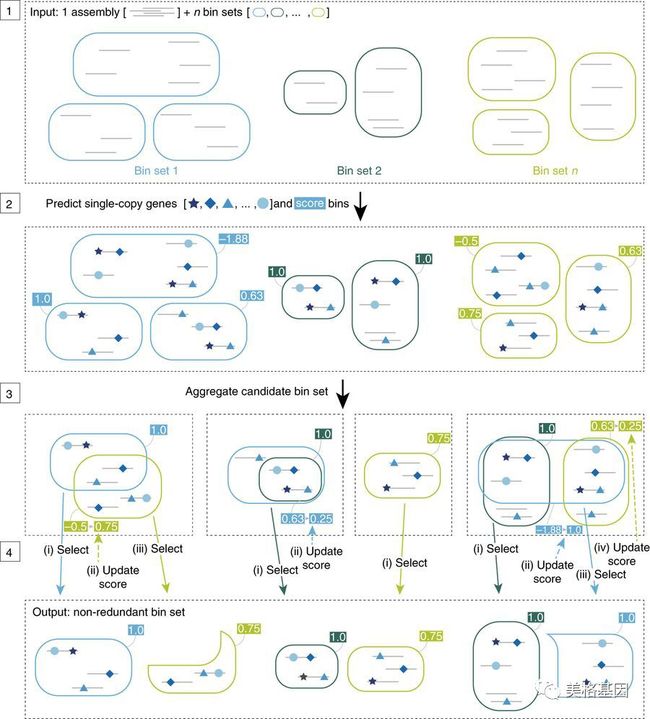
步骤1:DAS工具的输入文件包括拼接结果中的scaffolds序列(灰线表示)和来自不同binning工具得到的bins集合(相同颜色的圆角矩形表示由同一binning方法得到的bins);
步骤2:预测每个bins中scaffold的单拷贝基因(蓝色形状表示),并进行打分;
步骤3:在所有结果中,将相同的bins进行合并,作为这个bins的备选集合;
步骤4:迭代选择高分bins,并更新集合剩余部分候选bins的分数。
最终输出包括来自不同输入文件预测的非冗余高分bins。
2. DAS工具应用于模拟微生物群落
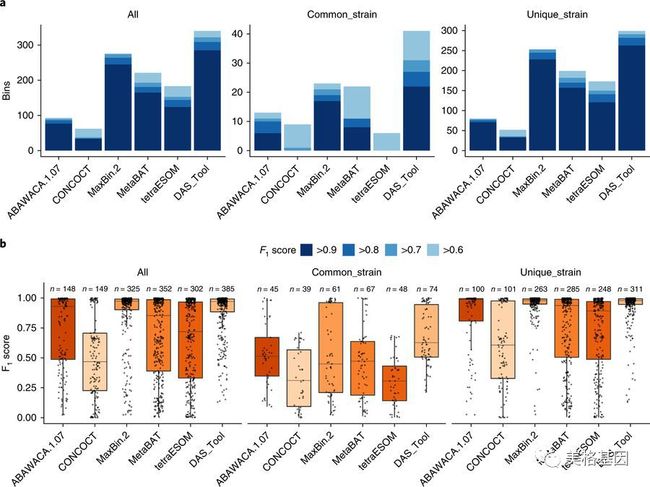
a, 每个方法重建的F1分数高于某个阈值的基因组的数量. F1得分越高, 重建的基因组与参照越相似. b, 所有bins的F1分数的分布. 在每个箱图上方给出每个方法重建bins的数目. 对所有参考基因组(ALL),具有菌株变异(common_strain; 与其他参考基因组的ANI(平均核苷酸相似性)≤95%)以及没有菌株变异(unique_strain; 与其他参考基因组的ANI > 95%ANI)的基因组三种情况分别计算度量。
以上结果展示,对于最具有挑战性的高复杂度样品,DAS工具可以比任何单个binning工具得到更多高质量的基因组,包括41个变异性菌株和299变异性菌株基因组。其次,Maxbin获得第二好的结果。DAS工具不仅重建更多数量基因组,其所有重建基因组的F1分数分布与最好的单分选工具(Maxbin)相比显示相等或更高的中值。
3. 应用于不同复杂程度的宏基因组数据

使用CheckM来评估所产生的bins的质量. 另外,使用ggKbase binning工具来分析人体肠道数据。鉴于人类肠道存在特定的细菌,这是合适的。 ggKbase工具没有用于其他样品分析,因为它们在含未知生物较多的系统中表现不佳。
由结果可以看出,在每个完整度水平下,DAS工具在3个生态系统中生成的高质量bins数量都是最多的。有趣的是,用作DAS工具输入的单个binning工具的性能在生态系统之间不同,而且它们都不是明显的赢家。对于单个binning工具来说,面对较低复杂度的人类肠道样品时,利用ggKbase可以产生最大数量的高质量bins,其次是MetaBAT。对于中等复杂性的渗漏油样品,单个binning工具中ABAWACA和MetaBAT产生的高质量基因组最多。而对于高复杂度的土壤数据,Maxbin则更具有优势。
4. 揭示了先前未报道的具有烃降解潜力的谱系
来自圣巴巴拉原油渗漏样品的宏基因组的binning结果揭示了三种基因组,其16S rRNA基因序列在SILVA数据库中没有找到相似序列(78.8、79.4和87.4%相似性)。这些重建基因组的完整性为95.6~89.6%。基于串联16个级联核糖体系统发育树,作者得出结论,其中2个基因组可能是来自2个新的门。第三个基因组更接近于WOR-3,可能是WOR-3的一个分支。
代谢通路分析显示3种基因组中存在编码烃降解酶的基因——包醛脱氢酶。此外,醇脱氢酶、醛铁氧还蛋白氧化还原酶和甲醇脱氢酶存在于一个基因组中,该基因组具有最高的完整性,推测具有烷烃和甲醇降解途径。
结果讨论
作者测试了一组当前可用的已发表的宏基因组学binning算法,以评估它们在应用于各种复杂性样本时的表现。然而,值得注意的是,在不同的环境系统类型中,甚至在同一生态系统的不同样本之间,这些都是工具的有效性都是可变的,并且没有单个binning工具一直是最有效的。而作者开发的DAS工具,几乎总是从复杂的宏基因组中提取出比任何单个binning工具更多的基因组。其次,由于Maxbin在本研究中的总体稳定表现,Maxbin结合2种或3种其他binning方法可以作为DAS工具的坚实基础。
从宏基因组重建的基因组的质量总是受到质疑。对于试图以基因组方式解决复杂生态系统的所有研究而言,不完善的基因组是一个挑战。不同的单一binning方法不仅产生不同数量的bins,而且基因组组成可能略有不同。这种可变性可以通过使用DAS工具来评估。在从每个binning工具中选取最好的bins时,DAS工具能够均衡单个binning工具之间的性能变化,从而增加回收的接近完整基因组的总数。因为它使用基于单拷贝基因的评分函数,所以它能够区分高质量和低质量的bins,并且通过使用适当的评分截止值,可以过滤出低质量的bins并控制megabins(多个bins混杂在一起)的数量。
总之,DAS工具可以集成ESOM等手动binning方法,也可以结合任何基于contig的binning算法的结果。因此,它具有高度的可扩展性,是可以利用和开发的binning工具。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外2600+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍末解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
