基于16S的细菌群落功能预测工具Tax4Fun2

基于16S的细菌群落功能预测工具Tax4Fun2

回想第一次接触微生物组分析时是2017年,那会儿主流的两个基于16S的细菌群落功能预测工具就是PICRUSt和Tax4Fun,均可以获得类似于宏基因组的功能丰度数据。不过当时来看Tax4Fun更推荐一些,Tax4Fun以 SILVA数据库的物种注释为基础(支持115、119和123三个版本,对应的程序最后一次更新是2015年,当时仅时隔2年),而PICRUSt以GreenGene数据库的物种注释为基础(最后更新是2013年,当时已时隔4年)。并且当时SILVA已经取代GreenGene成为了主流的16S细菌物种注释库,最后小编选择了Tax4Fun作分析。
其实再往后来看,这两个工具所依据的数据库也已经很老了。现在是2020年,SILVA都更新到138版本了,即便再使用Tax4Fun也很勉强(Tax4Fun最高仅支持SILVA123,但是再返回使用旧的数据库注释明显不是一个好选择)。
当然,生信工具也是与时俱进的。为了满足如今的所需,PICRUSt和 Tax4Fun都推出了全新版本,即PICRUSt2和Tax4Fun2。PICRUSt2的使用请参考隔壁的推文,本篇简介Tax4Fun2。
评估微生物群落的功能和冗余度是环境微生物学的主要挑战。Tax4Fun2是一个R程序包,可基于16S rRNA基因序列快速预测原核生物的功能谱和功能冗余。通过合并用户定义的、特定于栖息地的基因组信息,可以显著提高预测功能图谱的准确性和鲁棒性(Wemheuer et al, 2018)。与旧版Tax4Fun相比,Tax4Fun2的具有如下优点。
(1)不再局限于仅SILVA的特定版本注释的OTU丰度表,允许直接以OTU代表序列作为输入,通过与指定参考数据库的比对实现物种注释。除了Tax4Fun2提供的已构建好的参考集(相比之前大幅扩大),也允许我们提供自定义的参考集,使用非常灵活。
(2)侧重于原核数据,但也可以合并真核数据。
(3)提供了计算特定功能冗余的方法,对于预测特定功能在环境扰动期间丢失的可能性至关重要。
(4)精度和稳定性显著提升。
(5)目前Tax4Fun2仍处于不断的更新状态。
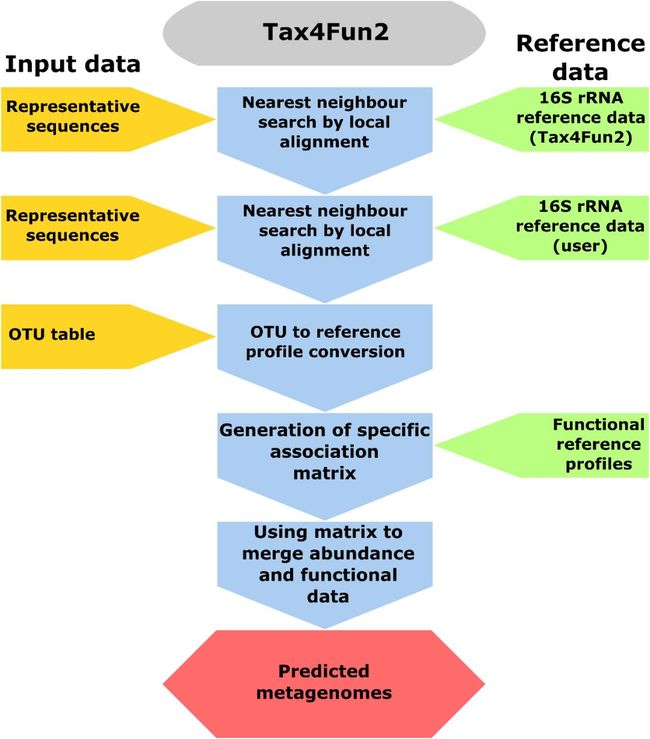
Tax4Fun2的工作流程。首先将16S rRNA基因序列与参考序列(可以为内置的,也允许使用用户自定义的)进行比对,以识别最近的邻居。根据最近邻居搜索的结果,汇总各样本的OTU丰度。一个关联矩阵(AM)包含在16S rRNA搜索中识别的参考功能概况。整合OTU丰度与AM功能谱,预测每个样品的宏基因组。
下文继续简介Tax4Fun2的使用。如果想了解关于旧版Tax4Fun的内容,可参考前文。
Tax4Fun2官方文档:https://github.com/bwemheu/Tax4Fun2
下文部分测试结果文件及R代码等,可在百度盘获取(提取码:kr9p):
https://pan.baidu.com/s/1nogKmaDSnexF34_WlfEWng
Tax4Fun2安装及配置
本篇的所有操作都是在linux平台下进行的,包括程序包的安装及测试过程。Windows或Mac平台下的方法可能存在不同,我未再尝试,大家若有需要可参考官方文档的说明。
参考自:https://github.com/bwemheu/Tax4Fun2
#下载 tax4fun2 程序包,shell 命令行
wget https://github.com/bwemheu/Tax4Fun2/releases/download/1.1.5/Tax4Fun2_1.1.5.tar.gz
然后打开R,手动安装Tax4Fun2程序包(它是一个R包)。
#安装 tax4fun2
install.packages(pkgs = 'Tax4Fun2_1.1.5.tar.gz', repos = NULL, source = TRUE)
#加载
library(Tax4Fun2)
随后,需要配置Tax4Fun2依赖环境,包括参考数据库,以及功能实现的其它R包或程序等。总体上来说安装无任何难度,都是命令行傻瓜式安装的。
加载Tax4Fun2包后,使用内部函数buildReferenceData()下载并构建默认的Tax4Fun2参考数据库,以及检查是否需要安装ape和seqinr包。此外,它还将测试用户PATH中是否存在blastn,为了确保下载成功,该功能还将使用md5sums自动检查下载数据的一致性。
#下载并构建默认的 Tax4Fun2 参考数据库
#path_to_working_directory='.',Tax4Fun2 默认在当前工作路径中构建库,如有需要,通过该参数指定位置
#use_force=FALSE,是否覆盖已有的文件夹,默认为 FALSE
#install_suggested_packages=TRUE,安装 ape 和 seqinr 包,默认为 TRUE
buildReferenceData(path_to_working_directory = '.', use_force = FALSE, install_suggested_packages = TRUE)
等待一会儿,执行成功后,可在当前工作路径下看到生成新路径“Tax4Fun2_ReferenceData_v2”,即为当前构建的Tax4Fun2参考数据库。在随后的功能预测环节,物种注释、基因组功能预测等,将通过这些库文件实现。

安装依赖,该函数将下载blast(v2.9.0)的当前最新版本,并将二进制程序放置在Tax4Fun2参考库路径中。另外,它还将测试ape和seqinr包是否可用。
#安装依赖
#path_to_working_directory,默认值为在当前工作路径中构建的库,如有需要,通过该参数指定位置
#use_force=FALSE,是否覆盖已有的文件夹,默认为 FALSE
#install_suggested_packages=TRUE,安装 ape 和 seqinr 包,默认为 TRUE
buildDependencies(path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
use_force = FALSE, install_suggested_packages = TRUE)
Tax4Fun2参考数据库路径“Tax4Fun2_ReferenceData_v2”中,新添加了blast程序。

Tax4Fun2预测细菌群落功能
接下来,测试Tax4Fun2的使用。
默认过程参考自:https://github.com/bwemheu/Tax4Fun2
1、测试数据集
使用官方提供的数据集测试。
#此命令将自动下载 Tax4Fun2 测试数据
#path_to_working_directory='.',自动下载到当前工作路径下,如有需要,通过该参数指定位置
getExampleData(path_to_working_directory = '.')
或者在以下链接手动获取(此外,我在网盘中也保存了一份)。
https://cloudstor.aarnet.edu.au/plus/s/4htgFinDIpzOuKK/download
通过如上命令获取后,这些测试数据自动下载到当前工作路径(以下除“Tax4Fun2_ReferenceData_v2”外,都是)。
接下来使用其中两个重要的文件。
“KELP_otu_table.txt”,就是常说的OTU丰度表(尽管它不是以OTU作为命名,但在习惯上,请允许我继续这样称呼),仅OTU ID及其在各样本(列)中的丰度信息,无OTU注释列。
“KELP_otus.fasta”,即各OTU的代表16S rRNA序列,fasta文件。

2、群落功能预测
第一步,物种注释
可以看到,和旧版Tax4Fun相比,新版Tax4Fun2中不再要求输入带SILVA特定版本注释的OTU丰度表。Tax4Fun2中,将使用提供的OTU代表16S rRNA序列,和已构建好的参考数据库中的已知物种的16S rRNA序列进行比对(默认调用blastn程序实现),根据相似性获得物种注释。
#物种注释
#指定 OTU 代表序列、Tax4Fun2 库的位置、参考数据库版本、序列比对(blastn)线程数等
runRefBlast(path_to_otus = 'KELP_otus.fasta', path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
path_to_temp_folder = 'Kelp_Ref99NR', database_mode = 'Ref99NR',
use_force = TRUE, num_threads = 4)
Tax4Fun2参考库“Tax4Fun2_ReferenceData_v2”中,有SILVA、NR库等可选(不妨自己点开看一下),上述使用了Ref99NR。
运行等待一会儿后,工作路径下获得“Kelp_Ref99NR”,存放了比对结果,输入序列和库中目标序列的最佳匹配。

第二步,KEGG Orthology(KO)功能及代谢途径丰度预测
在获得物种注释后,继续根据物种基因组的功能特征,结合OTU丰度表给出的物种丰度信息,推断群落功能丰度。
#预测群落功能
#指定 OTU 丰度表、Tax4Fun2 库的位置、参考数据库版本、上步的物种注释结果路径等
makeFunctionalPrediction(path_to_otu_table = 'KELP_otu_table.txt', path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
path_to_temp_folder = 'Kelp_Ref99NR', database_mode = 'Ref99NR',
normalize_by_copy_number = TRUE, min_identity_to_reference = 0.97, normalize_pathways = FALSE)
#或者
makeFunctionalPrediction(path_to_otu_table = 'KELP_otu_table.txt', path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
path_to_temp_folder = 'Kelp_Ref99NR', database_mode = 'Ref99NR',
normalize_by_copy_number = TRUE, min_identity_to_reference = 0.97, normalize_pathways = TRUE)
normalize_by_copy_number=TRUE,对于很多细菌而言,一个个体可能包含多条16S(多拷贝16S),因此推荐在原始OTU 16S rRNA丰度表的基础上,根据物种所含16S rRNA拷贝数对物种丰度进行标准化,得到校正16S rRNA拷贝数后的OTU丰度表后进行功能丰度映射。
normalize_pathways=FALSE,将每个KO的相对丰度关联到它所属的每个pathway;normalize_pathways=TRUE,每个KO的相对丰度平均分配给分配给它的所有pathway。
最后获得两个结果文件,如下所示。
“functional_prediction.txt”,预测到的功能基因在各样本中的相对丰度,功能基因映射到KO功能中(即KO第4级分类单元)。根据KO ID及功能描述信息,若有感兴趣的,可在KEGG官网(https://www.genome.jp/kegg/pathway.html)查询获得更多细节。
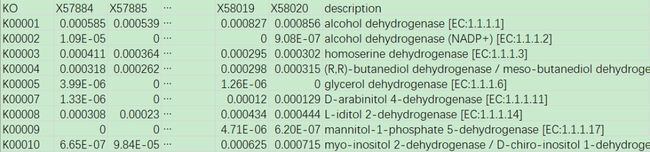
“pathway_prediction.txt”,将功能基因丰度映射到其所属的代谢途径(KEGG pathway,即KO第3级分类单元)中,获得各样本中各pathway的相对丰度。在这里也能看到Tax4Fun2和早期版本的Tax4Fun相比的一个改进点,就是将这些pathway所属的更高级KO分类单元也给了出来,方便我们归类分析;而早期版本的Tax4Fun中是没有这些高级分类信息的(通常还需手动添加,我在前文Tax4Fun教程中也提到了手动添加到方法)。

3、统计分析
获得群落功能丰度表后,就可以按照OTU丰度表的统计分析方法,去执行类似的分析了。这点可以找一些文献作参考,看别人是怎样做的。例如,首先计算特定功能丰度在组间的显著性,获得组间差异显著的功能,然后再从数据库官网上查询该功能的细节,解释生物学现象等。
Tax4Fun2的其它实用功能
除了基于16S rRNA丰度的细菌群落功能预测,Tax4Fun2还提供了其它很多实用功能。上述我们下载了Tax4Fun2的示例数据。除了OTU丰度表以及OTU代表序列外,还有很多其它文件,继续使用这些示例数据作展示。
同样地,参考自:https://github.com/bwemheu/Tax4Fun2
构建自定义参考集
Tax4Fun2还允许我们自定义构建参考集,充分体现了它的灵活性。
1、提取SSU序列
指定一个单一物种基因组序列fasta文件,或者包含多个基因组fasta文件的文件夹。基因组序列中必须含有至少一个拷贝的16S rRNA基因序列,运行命令可获取其中的16S rRNA序列,并删除“空”基因组。
#提取 SSU 序列,16S rRNA
#给定单个基因组
extractSSU(genome_file = 'OneProkaryoticGenome.fasta', file_extension = 'fasta',
path_to_reference_data = './Tax4Fun2_ReferenceData_v2')
#给定多个基因组
extractSSU(genome_folder = 'MoreProkaryoticGenomes', file_extension = 'fasta',
path_to_reference_data = './Tax4Fun2_ReferenceData_v2')
2、为原核基因组分配功能
类似地,指定一个单一物种基因组序列fasta文件,或者包含多个基因组fasta文件的文件夹。Tax4Fun2将调用prodigal、diamond blast等程序,实现类似原核物种基因组功能注释的过程。
#为原核基因组分配功能
#给定单个基因组
assignFunction(genome_file = 'OneProkaryoticGenome.fasta', file_extension = 'fasta',
path_to_reference_data = './Tax4Fun2_ReferenceData_v2', num_of_threads = 1, fast = TRUE)
#给定多个基因组
assignFunction(genome_folder = 'MoreProkaryoticGenomes/', file_extension = 'fasta',
path_to_reference_data = './Tax4Fun2_ReferenceData_v2', num_of_threads = 1, fast = TRUE)
3、生成参考数据
提取SSU序列以及完成基因组功能分配后,对于每个给定的基因组至少获得两个文件:一个记录SSU序列,一个记录功能注释概况。最后指定这些文件所在的文件夹并提供正确的文件扩展名,生成最终数据集。
#生成参考数据,文档中提供了 3 种方法
#推荐选择第二种,该命令包含一个 uclust 聚类步骤,可消除数据中的冗余
# 1) Generate user-defined reference data without uclust from a single genome
generateUserData(path_to_reference_data = './Tax4Fun2_ReferenceData_v2', path_to_user_data = '.',
name_of_user_data = 'User_Ref0', SSU_file_extension = '_16SrRNA.ffn', KEGG_file_extension = '_funPro.txt')
# 2) Generate user-defined reference data without uclust
generateUserData(path_to_reference_data = './Tax4Fun2_ReferenceData_v2', path_to_user_data = 'MoreProkaryoticGenomes',
name_of_user_data = 'User_Ref1', SSU_file_extension = '_16SrRNA.ffn', KEGG_file_extension = '_funPro.txt')
# 3) Generate user-defined reference data without uclust
generateUserDataByClustering(path_to_reference_data = './Tax4Fun2_ReferenceData_v2', path_to_user_data = 'MoreProkaryoticGenomes',
name_of_user_data = 'User_Ref2', SSU_file_extension = '_16SrRNA.ffn', KEGG_file_extension = '_funPro.txt', use_force = TRUE)
计算(多)功能冗余指数(functional redundancy indices,FRI)
计算KEGG功能的系统发育分布(高FRI->高冗余度,低FRI ->低冗余度,并可能随群落变化而丢失)。
##计算(多)功能冗余指数(FRIs)
#OTU 代表序列与数据库参考序列的 blast
runRefBlast(path_to_otus = 'Water_otus.fna', path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
path_to_temp_folder = 'Water_Ref99NR', database_mode = 'Ref99NR', use_force = TRUE, num_threads = 4)
#计算 FRIs 值
calculateFunctionalRedundancy(path_to_otu_table = 'Water_otu_table.txt', path_to_reference_data = './Tax4Fun2_ReferenceData_v2',
path_to_temp_folder = 'Water_Ref99NR', database_mode = 'Ref99NR', min_identity_to_reference = 0.97)
参考文献
Wemheuer F, Taylor J A, Daniel R, et al. Tax4Fun2: a R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. bioRxiv, 2018.
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读