R语言绘制带聚类树的堆叠柱形图

R语言绘制带聚类树的堆叠柱形图

聚类树与柱形图结合,即可反映样本或分组间的相似性,又能展示样本内的元素组成信息。
例如下图是一个在扩增子测序微生物群落分析中常见的统计图类型,在测序公司给的报告中通常都有这么一张图。聚类树表示了各样本或分组之间,物种丰度组成的相似性或相异程度;柱形图就是常见的物种堆叠柱形图,常用于表示代表性的门纲目科属等丰度,如下图中即展示了各样本中丰度排名前10的细菌门的丰度信息。
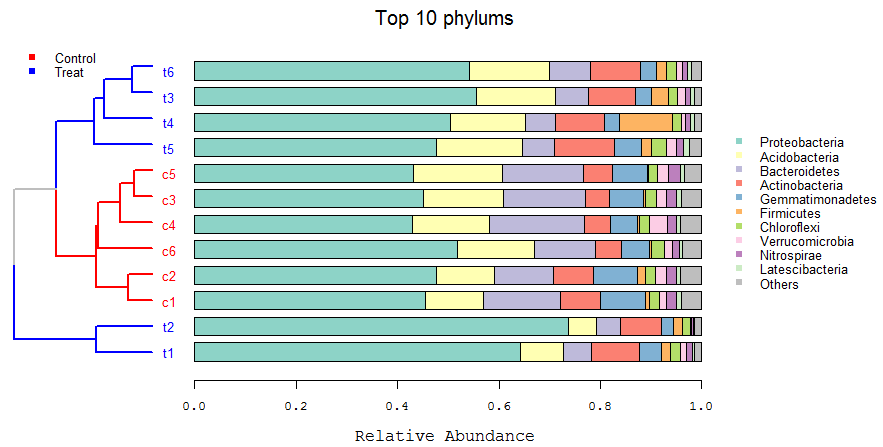
很容易看出,该图主体由两部分组成,聚类树+堆叠柱形图。
本篇分享怎样在R语言中绘制。
示例数据,R代码等的百度盘链接:
https://pan.baidu.com/s/1mcn3XCma9_0C2LpDVytYsg
作图示例文件
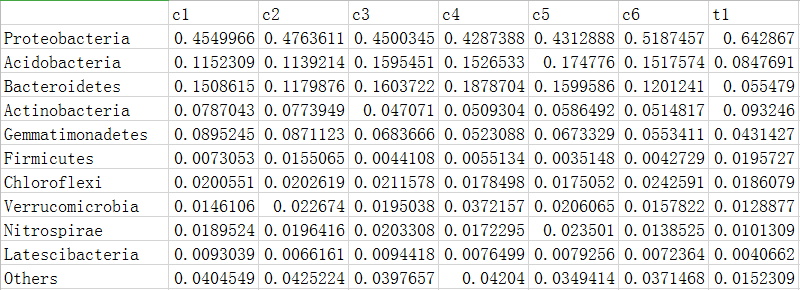
文件“phylum_top10.txt”由16S高通量测序所得的物种丰度表计算得到,记录了丰度排名top10的主要细菌类群(在门水平统计,行)在各样本(12个样本,列)中的丰度信息。Top10丰度外的类群合并为Others。
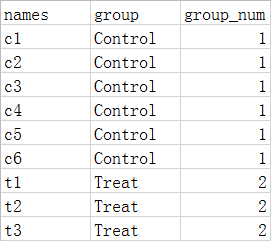
“group.txt”为12个样本的分组信息。
通过这两个文件绘制带聚类树的堆叠柱形图。
R语言作图
首先进行层次聚类,获得代表样本间物种组成相似性的聚类树,这一步很简单。
#层次聚类
#读取 OTU 丰度表
dat <- read.delim('phylum_top10.txt', row.names = 1, sep = '\t', head = TRUE, check.names = FALSE)
#计算样本间距离,以群落分析中常用的 Bray-curtis 距离为例
dis_bray <- vegan::vegdist(t(dat), method = 'bray')
#层次聚类,以 UPGMA 为例
tree <- hclust(dis_bray, method = 'average')
tree
plot(tree)
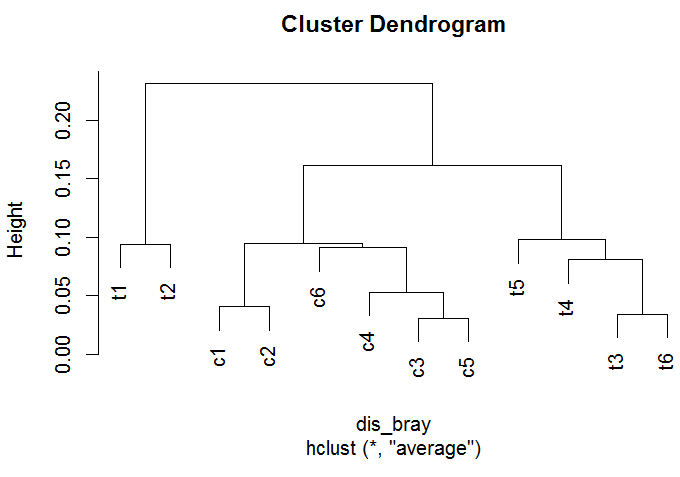
接下来就是对聚类树进行调整。
具体做法,首先定义一个画板,将聚类树放在画板的左侧,并按样本的已知分组信息给分支上色。聚类树的一些常见的可视化调整方法,可参考前文。
##聚类树绘制
#样本分组颜色、名称等
group <- read.delim('group.txt', row.names = 1, sep = '\t', head = TRUE, check.names = FALSE, stringsAsFactors = FALSE)
grp <- group[2]
group_col <- c('red', 'blue')
names(group_col) <- c('1', '2')
group_name <- c('Control', 'Treat')
#样本分组标签
layout(t(c(1, 2, 2, 2, 3)))
par(mar = c(5, 2, 5, 0))
plot(0, type = 'n', xaxt = 'n', yaxt = 'n', frame.plot = FALSE, xlab = '', ylab = '',
xlim = c(-max(tree$height), 0), ylim = c(0, length(tree$order)))
legend('topleft', legend = group_name, pch = 15, col = group_col, bty = 'n', cex = 1)
#聚类树绘制,按分组给分支上色
treeline <- function(pos1, pos2, height, col1, col2) {
meanpos = (pos1[1] + pos2[1]) / 2
segments(y0 = pos1[1] - 0.4, x0 = -pos1[2], y1 = pos1[1] - 0.4, x1 = -height, col = col1,lwd = 2)
segments(y0 = pos1[1] - 0.4, x0 = -height, y1 = meanpos - 0.4, x1 = -height, col = col1,lwd = 2)
segments(y0 = meanpos - 0.4, x0 = -height, y1 = pos2[1] - 0.4, x1 = -height, col = col2,lwd = 2)
segments(y0 = pos2[1] - 0.4, x0 = -height, y1 = pos2[1] - 0.4, x1 = -pos2[2], col = col2,lwd = 2)
}
meanpos = matrix(rep(0, 2 * length(tree$order)), ncol = 2)
meancol = rep(0, length(tree$order))
for (step in 1:nrow(tree$merge)) {
if(tree$merge[step, 1] < 0){
pos1 <- c(which(tree$order == -tree$merge[step, 1]), 0)
col1 <- group_col[as.character(grp[tree$labels[-tree$merge[step, 1]],1])]
} else {
pos1 <- meanpos[tree$merge[step, 1], ]
col1 <- meancol[tree$merge[step, 1]]
}
if (tree$merge[step, 2] < 0) {
pos2 <- c(which(tree$order == -tree$merge[step, 2]), 0)
col2 <- group_col[as.character(grp[tree$labels[-tree$merge[step, 2]],1])]
} else {
pos2 <- meanpos[tree$merge[step, 2], ]
col2 <- meancol[tree$merge[step, 2]]
}
height <- tree$height[step]
treeline(pos1, pos2, height, col1, col2)
meanpos[step, ] <- c((pos1[1] + pos2[1]) / 2, height)
if (col1 == col2) meancol[step] <- col1 else meancol[step] <- 'grey'
}

按分组给聚类树的分支或簇标记了颜色,放置在画板左侧的一小部分区域。
对于右侧区域,放置堆叠柱形图,用以展示top10丰度的细菌门在各样本中的丰度信息。堆叠柱形图单独的画法,可参考该文。
##堆叠柱形图
#样本顺序调整为和聚类树中的顺序一致
dat <- dat[ ,tree$order]
#物种颜色设置
phylum_color <- c('#8DD3C7', '#FFFFB3', '#BEBADA', '#FB8072', '#80B1D3', '#FDB462', '#B3DE69', '#FCCDE5', '#BC80BD', '#CCEBC5', 'gray')
names(phylum_color) <- rownames(dat)
#堆叠柱形图
par(mar = c(5, 2, 5, 0))
bar <- barplot(as.matrix(dat), col = phylum_color, space = 0.4, width = 0.7, cex.axis = 1, horiz = TRUE, cex.lab = 1.2,
xlab = 'Relative Abundance', yaxt = 'n', las = 1, ylim = c(0, ncol(dat)), family = 'mono')
# 注意:百度云链接中的代码将tree$order打错为test_order,请在运行中报错自行修改
mtext('Top 10 phylums', side = 3, line = 1, cex = 1)
text(x = -0.05, y = bar, labels = colnames(dat), col = group_col[group[tree$order, 2]], xpd = TRUE)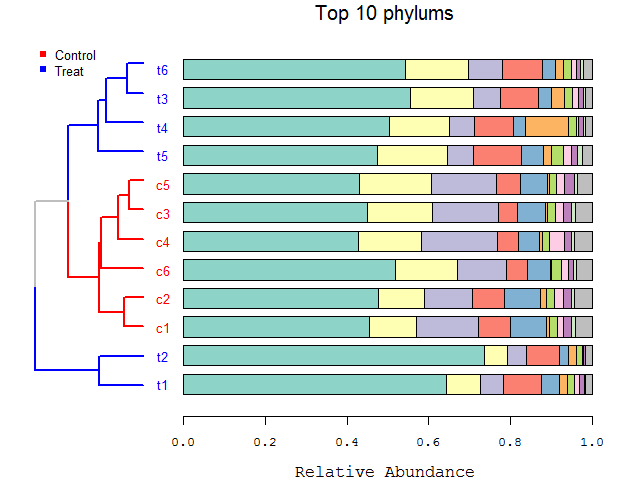
首先按聚类树中样本的顺序重新调整丰度表中样本的顺序,以保持二者能够对应,并定义颜色属性。之后绘制堆叠柱形图,放置在画板右侧区域。
最后在最右侧添加柱形图图例,哪些颜色代表了哪种细菌类群。
#柱形图图例
par(mar = c(5, 1, 5, 0))
plot(0, type = 'n', xaxt = 'n', yaxt = 'n', bty = 'n', xlab = '', ylab = '')
legend('left', pch = 15, col = phylum_color, legend = names(phylum_color), bty = 'n', cex = 1)
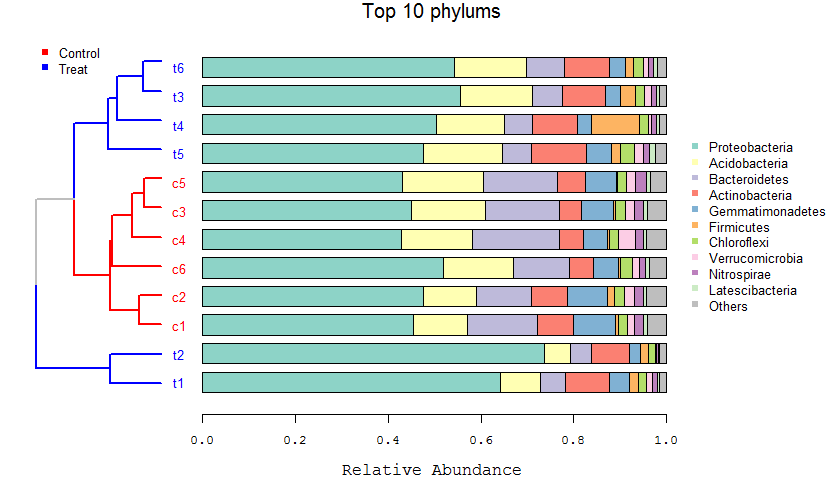
这样这种带聚类的堆叠柱形图就搞定了。
其它说明
如果觉得使用R组合两张图比较麻烦,特别是细节部分不容易调整,不妨分别绘制聚类树和堆叠柱形图,然后使用AI、PS等工具将二者拼合在一起,可能相对方便许多。再略加修整,也会更好看。
猜你喜欢
10000+:菌群分析 宝宝与猫狗 梅毒狂想曲 提DNA发Nature Cell专刊 肠道指挥大脑
系列教程:微生物组入门 Biostar 微生物组 宏基因组
专业技能:学术图表 高分文章 生信宝典 不可或缺的人
一文读懂:宏基因组 寄生虫益处 进化树
必备技能:提问 搜索 Endnote
文献阅读 热心肠 SemanticScholar Geenmedical
扩增子分析:图表解读 分析流程 统计绘图
16S功能预测 PICRUSt FAPROTAX Bugbase Tax4Fun
在线工具:16S预测培养基 生信绘图
科研经验:云笔记 云协作 公众号
编程模板: Shell R Perl
生物科普: 肠道细菌 人体上的生命 生命大跃进 细胞暗战 人体奥秘
写在后面
为鼓励读者交流、快速解决科研困难,我们建立了“宏基因组”专业讨论群,目前己有国内外5000+ 一线科研人员加入。参与讨论,获得专业解答,欢迎分享此文至朋友圈,并扫码加主编好友带你入群,务必备注“姓名-单位-研究方向-职称/年级”。PI请明示身份,另有海内外微生物相关PI群供大佬合作交流。技术问题寻求帮助,首先阅读《如何优雅的提问》学习解决问题思路,仍未解决群内讨论,问题不私聊,帮助同行。

学习16S扩增子、宏基因组科研思路和分析实战,关注“宏基因组”
 点击阅读原文,跳转最新文章目录阅读
点击阅读原文,跳转最新文章目录阅读