微生物绝对定量研究知多少
目前细菌群落研究常用的技术是16S相对定量扩增子测序,而最近越来越多的研究者开始采用绝对丰度(即菌群的实际数量)对细菌群落进行绝对定量研究,今天小编就微生物绝对定量研究的一些常见问题在此总结一下。
问答1
绝对定量相对于常规16S相对定量扩增子测序最大的优势是什么?
常规16S扩增子测序方法能解析样本中的物种组成和其相对丰度信息,因此对微生态研究具有重要的促进作用。然而我们需要注意一点,那就是相对丰度并不能反映样本每种微生物的真实数量和组间样本的真实差异,这是为什么呢?这是因为相对丰度分析忽略了不同样本之间总体微生物量存在的现实差异,造成了分析结果的偏差,而绝对定量分析则是计量样本每种微生物的16S基因拷贝数或者数目,从而实现绝对定量,因此相对于常规16S相对定量扩增子测序,绝对定量分析能反映样本每种微生物的真实数量和组间样本的真实差异,因此绝对定量分析相对于相对定量分析更能反映样本细菌群落的真实变化,是进行微生态研究的首选。
支持文献1:Quantitative microbiome profiling links gut community variation to microbial load. Nature. 2017.551(7681):507-511.(该文章链接,请点击:Nature:要想真正研究宿主-肠道微生物的相互作用,必须将相对定量变成绝对定量)
支持证据:
为了证明绝对定量数据(QMP)在临床研究中的潜在作用,本研究对29例克罗恩病患者和健康人的样本进行绝对定量数据分析,流式细胞仪分析表明,克罗恩病患者粪便样品中的细胞数比健康对照组低3倍(图4a)。
发现只在应用相对数据RMP分析时,拟杆菌Bacteroides与克罗恩病相关(图4b左),仅在使用绝对定量数据QMP时,普雷沃菌属Prevotella与克罗恩病相关(图4b右)。
以上这些观察强调了相对数据(RMP)分析的局限性:来源于相对数据(RMP)分析的一个错误结果解释可能提示拟杆菌Bacteroides在克罗恩病的发病或发展中的假因果关系,为什么会出现这种假因果关系,这是因为相对定量分析忽视了健康对照和克罗恩病患者肠道微生物总丰度的巨大差异(差3倍),通过人为抽平处理认为样本间微生物总丰度一致,从而造成上述错误,因此本文认为绝对定量数据(QMP)分析鉴定到的微生物总丰度降低才是引起克罗恩病患者肠道微生物结构发生改变的根本原因。本文认为目前大多数对于肠道微生物菌群结构的研究并不能体现出具体微生物数量改变对宿主带来的影响,这些相对丰度研究忽略了总体微生物丰度本身的改变可能是引起宿主产生疾病的主要原因这一可能性。所以,为了真正体现宿主-微生物群的相互作用,微生物群的研究必须将相对丰度变成绝对丰度。
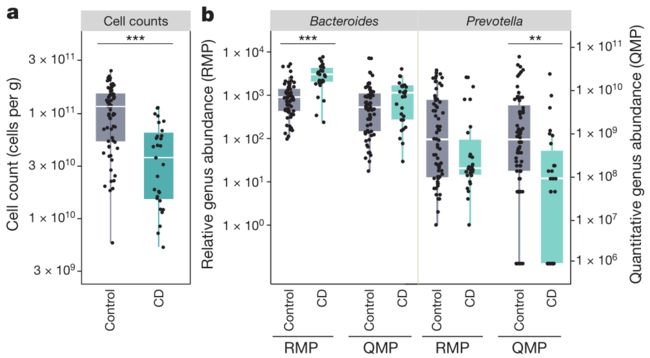
图4. 克罗恩病的微生物数量变化
支持文献2:Absolute quantification of microbial taxon abundances. The ISME Journal. 2017.11(2):584-587. (该文章链接,请点击:The ISME Journal:为什么微生物相对定量不能代替绝对定量)
支持证据:
本研究对核试验反应堆上运行的冷却水回路系统进行两次40天调查(survey1和survey2),包括冷却水系统没有运行的时间段(control),冷却水系统启动阶段(start-up),冷却水系统稳态运行阶段(operation)。
仔细检查了两个微生物分支betI-A分支和bacI-A分支的释相对丰度和绝对丰度的时间轨迹时间轨迹(图2)。结果检测到两个主要差异:1):在群落成长期间,比如survey 1的start-up和早期 operation阶段,绝对丰度有一个明确的过渡,显示出bacI-A分支的生长和衰变;相反,在生长和衰变过程中,bacI-A分支的相对丰度则始终保持相对恒定,没有任何过渡。2):在survey2的operation后半部分,相对丰度曲线显示从±40%增加到90%以上,暗示了betI-A分支的生长;相反,如果看绝对丰度,那么这个分支就没有明显的活跃生长,其细胞密度从未显著超过start-up阶段结束时观察到的最大密度。
本文认为微生物某一类群的富集(相对丰度比例的增加)不一定真正与相应微生物类群生长(绝对丰度的增加)有关,可能是其它微生物类群的衰退导致了其在群落结构中相对丰度比例的增长。
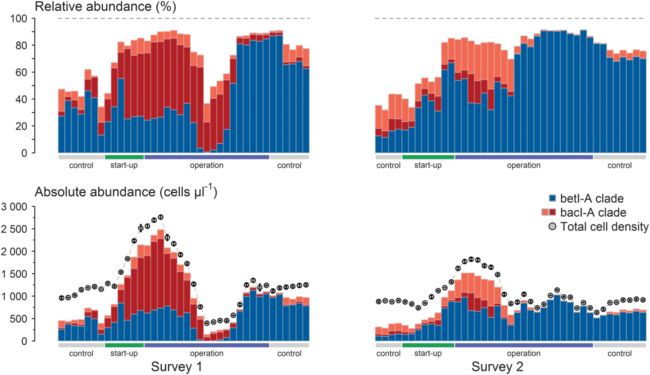
图2 对在核试验反应堆上运行的二级冷却水回路进行两次时间分离的40天调查期间,2个最丰富的微生物淡水分支(bacI-A(OTU2,红色;OTU3,橙色)和betI-A(OTU1,蓝色)的时间动态图。top panel显示从扩增子测序数据中推断的相对丰度(in%),bottom panel显示绝对OTU丰度(单位为细胞/μl), 圆形标签表示微生物群落的总细胞密度(单位为细胞/μl±s.d)。最下方灰色区域表示冷却水系统没有运行的时间段(控制阶段),绿色区域表示启动,蓝色区域表示稳态运行。
问答2
既然绝对丰度更准确,那么联合常规16S相对定量扩增子测序和总细菌qPCR获得的绝对丰度可靠吗?
qPCR是定量物种绝对丰度的金标准,但是针对存在于样品中的每种细菌开发一套qPCR检测方法显然是不切实际的,因此很多老师经常联合常规16S相对定量扩增子测序(获取每个物种的相对丰度)和总细菌qPCR(获取总细菌载量)来获得每个物种的绝对丰度,总体而言,基于这两种技术联合推断的特定物种的绝对丰度在某种程度上提供了一个机会来评估细菌物种的绝对丰度,但是有研究表明这种联合只适合高丰度物种(物种的相对丰度大于10%)的绝对丰度推断,当物种的相对丰度较低时,这种联合分析推断的绝对丰度估计值很容易出错,并且可能在从低细菌丰度增殖时期和后期收缩到低丰度的过程中可能会歪曲单个物种的动力学。
支持文献1:Complementing 16S rRNA Gene Amplicon Sequencing with Total Bacterial Load To Infer Absolute Species Concentrations in the Vaginal Microbiome. mSystems. 2020.5(2):e00777-19.(该文章链接,请点击:相对丰度会歪曲实际丰度,联合16S扩增子测序和总菌qPCR获得的绝对丰度可靠吗?)
支持证据:
本研究分析了来自20名有细菌性生殖道病史的女性的1320份样本,通过获取物种相对丰度(通过16S扩增子测序获得)和总细菌载量(通过细菌16S通用引物 qPCR获得)的乘积来推断每个细菌的绝对丰度,同时通过与物种靶向qPCR测定的阴道微生物组中7个关键物种的绝对丰度相比较来验证上述推断的绝对丰度估计值的准确性。
单一物种当用qPCR检测时,结果变化和扩增子测序检测结果变化不同,例如,对于图1中所示的参与者,A. vaginae的绝对丰度在第17天(h 415)突然增加,但是相对丰度直到第28天(h 671),才显示出突然增加。从第0天到第7天(h 168),受试者接受甲硝唑治疗细菌性阴道炎(BV):qPCR显示BV相关物种的绝对丰度呈指数下降;然而,扩增子测序则显示受试者经过治疗更迅速地向Lactobacillus iners转化。另外,扩增子测序的相对丰度也无法捕获细菌的低水平定植,例如扩增子测序在第6至11天(h 150和261)不能发现Gardnerella vaginalis的定殖,而靶向qPCR则可以发现Gardnerella vaginalis的定殖。综上,纵向剖面的比较再次说明相对丰度和绝对丰度之间确实存在很大差异。
本研究将推断的绝对丰度值与七个关键物种的靶向qPCR所测量的绝对丰度进行了比较,对于每个物种,在大多数样品中推断出的细菌绝对丰度与qPCR所测量的绝对丰度很类似(图1c;图2中的虚线)。在许多情况下,对于大多数物种,没有明显的极端不一致(图2a)。然而,对于某些物种,如Megasphaera和BVAB2,推断的细菌绝对丰度始终高于qPCR所测量的绝对丰度一个数量级(图2b)。在一组样本中,对于所有物种,推断的细菌绝对丰度为零,而qPCR水平为正,导致推断的细菌绝对丰度和qPCR所测量的绝对丰度之间的严重不一致:这通常在低绝对丰度情况下出现(图2)。
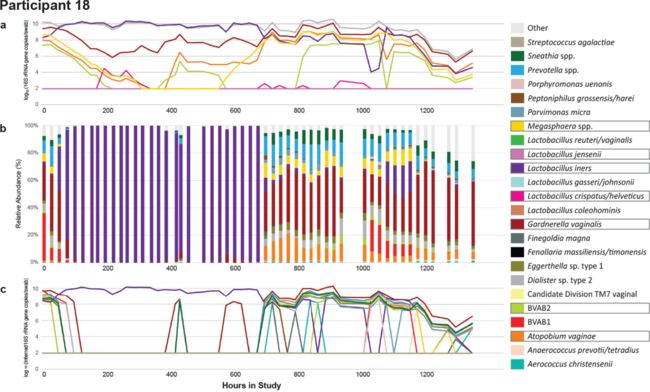
图1 参与者生殖道生态位的复杂细菌动力学。对一名18岁女性每日采集的生殖道拭子样本进行分析:(a)针对七个特定物种的定向qPCR;(b)16S扩增子测序;(c)相对丰度超过1%的物种的绝对丰度推断值。方框表示已使用qPCR对特定菌进行了检测。
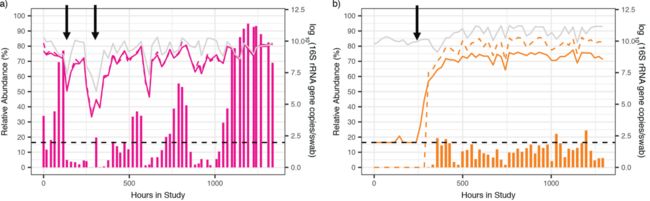
图2 由于细菌总负荷的变化,相对丰度估计可能会歪曲实际丰度。两名受试者中两种不同物种的物种特异性特征的示例:crispatus,受试者06(a)和Megasphaera,受试者17(b)。垂直条显示相对丰度(%,左y轴),实线表示qPCR测量的绝对丰度,灰色线表示细菌总负荷,虚线表示推断丰度(右y轴)。黑色虚线表示qPCR数据的检测阈值。箭头表示相对丰度变化与绝对丰度变化不一致的时间点,这通常发生在细菌负荷急剧变化或相对丰度较低时。
问答3
绝对定量分析的计算单位是用16S copies/g样本还是用16S copies/ng DNA,哪个更准确?
在实验条件允许情况下,建议采用样本水平(采用16S copies/g粪便、16S copies/g土壤、16S copies/mL 水作单位)进行后续群落分析,其结果也更符合实验预期,天昊生物丰富的16S扩增子绝对定量测序项目经验表明样本水平的聚类效果要比DNA水平(16S copies/ng DNA)好,往往组间差异增大,组内差异减小(见下图)(小贴士:a,注意记录抽提DNA时每个样本所用量(即用了多少g 土壤/粪便或mL 水);b,记录每个样本抽提获得的DNA总量(即抽提出多少ng DNA),以上数据可以将测序获得的16S copies/ ng DNA数值转化成发表文章时所用的 16S copies/g样本数值)。

问答4
绝对定量分析得到的16S copies/g样本数据和真实的微生物数目一致吗?
无论是物种靶向qPCR绝对定量或者基于内标法的绝对定量测序得到的每克样本的16S copies数据都与每克样本中所包含的微生物细胞数是有差异的,原因是因为大部分物种中的16S RNA基因会有多个拷贝,并且不同物种的16S RNA基因拷贝数不同,这就需要对得到的16S copies数据进行拷贝数矫正。rrnDB 数据库,全称ribosomal RNA operons (rrn) DataBase,是一个收集了NCBI全基因组数据中细菌和古菌的16S基因拷贝数的数据库,但是目前rrnDB数据库收录的数据有限,无法得到所有物种的拷贝数信息,而天昊生物基于创新算法可以得到所有物种的拷贝数信息,从而提取OTU对应的16S基因拷贝数信息,矫正OTU拷贝数,经过矫正后的OTU绝对拷贝数更接近微生物物种的细胞数,基于矫正过的16S基因拷贝数数据进行后续的生信分析结果更加真实可靠。
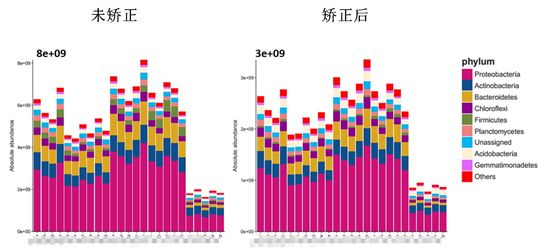
问答5
绝对定量分析得到的结果在和代谢组联合分析方面有什么优势?
通过绝对定量分析可以得到物种的绝对丰度,而目前代谢组中的靶向代谢组和全靶代谢组得到的是代谢物的绝对浓度,但是目前微生物组和代谢组的联合分析往往基于物种相对丰度和代谢物绝对浓度来进行相关性分析,这显然会造成错误的结论,因此强烈推荐基于物种绝对丰度和代谢物绝对浓度进行后续的相关性分析,这样得到的结果显然更加真实可靠。
各位看官读完上述文章如果有想做微生物绝对定量的想法,
那就请联系天昊生物吧!
天昊生物是国内唯一一家提供 “微生物16S扩增子绝对定量测序”技术的服务商。
![]()
天昊16S扩增子绝对定量测序技术简介
该技术是一种将qPCR绝对定量技术和常规16S扩增子测序技术合二为一的技术,该方法通过向样品DNA中添加拷贝数已知的内标序列DNA ,微生物DNA和内标序列DNA一起进行PCR扩增,然后一起进行16S扩增子文库构建、测序,再根据内标序列的16S扩增子序列数及其绝对拷贝数绘制标准曲线,计算出样品中OTU代表序列对应物种16S rRNA基因绝对拷贝数。该技术不但可以进行Alpha多样性分析、群落组成分析、Beta多样性分析、指标和微生物相关性分析等常规16S扩增子测序分析,关键可以解析样本中总细菌的绝对拷贝数,还可以解析样本中每个物种的绝对拷贝数,因而对微生态学内许多悬而未决的问题具有进一步阐明的潜力。此外,该技术进行细菌拷贝数定量时,构建标准曲线的内标和样本DNA是在同一个样本孔中一起进行PCR反应,所以PCR反应效率相同,因此校正了腐殖酸对PCR的影响,避免了腐殖酸等PCR抑制物对样品细菌16S拷贝数定量的影响,因此针对土壤、水体和淤泥等环境样本,天昊生物16S扩增子绝对定量测序技术计算得到的细菌16S拷贝数相对于qPCR更准确。
![]()
天昊16S扩增子绝对定量测序技术
应用情况
目前天昊微生物16S扩增子绝对定量测序技术平台已经完成项目百余个,合作单位包括中国科学院微生物研究所、中国科学院南京土壤研究所、中国科学院水生生物研究所、同济大学环境科学与工程学院、厦门大学环境与生态学院、中国农业大学、南京农业大学、东北农业大学、重庆市农业科学院、盐城工学院、南京财经大学、南京中医药大学、武汉大学中南医院、新疆医科大学公共卫生学院、山东大学齐鲁医院等多个单位,覆盖环境土壤微生物,环境水体微生物和医学肠道微生物等多个领域,利用该技术的项目文章成功发表在环境科学期刊《Science of the Total Environment》(IF= 6.551)和应用化学1区期刊《Carbohydrate Polymers》(IF=7.182)上,目前该技术因其创新性、准确性和稳定性受到客户的广泛好评! 天昊生物目前是国内唯一一家提供 “微生物16S扩增子绝对定量测序”技术的服务商,热烈欢迎各位老师与我们交流沟通!
想了解更多“天昊微生物16S扩增子绝对定量测序技术”内容,请点击链接:
1、喜讯!天昊生物16S扩增子绝对定量测序项目文章再次登陆《Science of the Total Environment》;
2、又一篇!天昊生物微生物16S扩增子绝对定量测序技术再发好文;
3、祝贺!天昊生物16S扩增子绝对定量测序技术助力客户登陆Science of the Total Environment;
咨询沟通也可联系
18964693703(微信同号)

创新基因科技,成就科学梦想