Samtools安装与使用
由于二代测序中普遍采取短读长(50~150bp)的测序策略,在后续分析的流程中需要使用比对软件将reads片段匹配到参考基因组中从而产生比对/匹配文件,进而用于后续流程的分析。
Samtools是一个用来处理SAM/BAM(SAM的二进制格式,用于压缩空间)格式的比对文件的工具,它能够输入和输出SAM(sequence alignment/map:序列比对)格式的文件,对其进行排序、合并、建立索引等处理。Samtools于2009年由Li Heng 发表在期刊BIOINFORMATICS上,被广泛地应用并整合在二代测序分析流程中,至今已超过2w+的引用率。

doi:10.1093/bioinformatics/btp352
一.安装
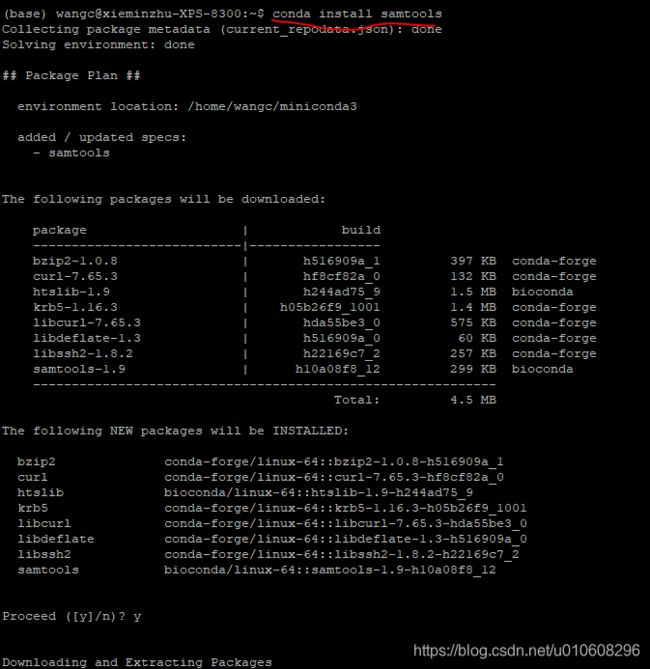
conda install samtools安装成功后,在终端输入 samtools ,出现如下界面后,表示安装成
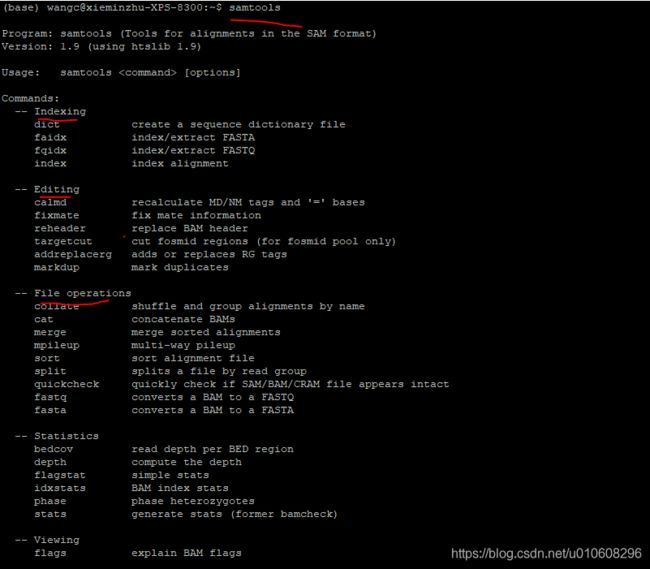
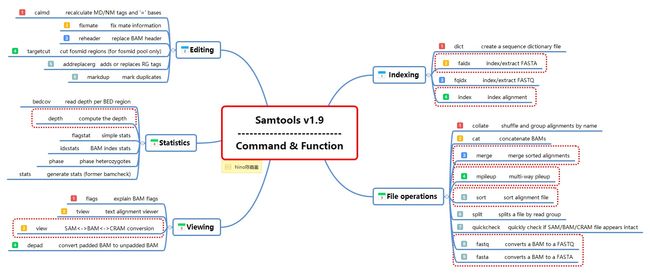
分为5类命令块:Indexing, Editing, File operations, Statistics, Viewing,
二.功能与命令(常用)
(1)faidx :index/extract FASTA
本命令对参考基因组(如hg19等)的fasta文件建立索引文件, 生成的文件以.fai后缀结尾。
$ samtools faidx ref.fasta(2)sort: sort alignment file
本命令对bam文件中的序列进行排序,默认下是按序列在fasta文件中的顺序(即header)和序列从左往右的位点排序。
$ samtools sort [-T out.prefix][-@ threads][-m maxMem][-o out.sorted.bam][in.bam]- -TPREFIX:把临时文件写到 PREFIX.bam. 里面。这个参数是必须的。
- -@INT:设置排序和压缩的线程数,默认是单线程。
- -mINT:设置每个线程的最大内存,以 bytes 为单位设定或者添加一个 K, M, 或 G 后缀。
- -oFILE: 将最后排好序后输出到 FILE 中,而不是标准输出。
(3)merge: merge sorted alignments
本命令将多个排好序的bam比对文件进行合并,产生一个排好序的bam输出文件(合并后的文件不需要再进行sort),这个文件含有所有的输入记录,并且保留了他们原来的顺序。
$ samtools merge [out.bam][in1.bam][in2.bam][in3.bam](4)index:index alignment
本命令对bam文件建立索引并产生后缀为.bai的文件,用于快速的随机处理。很多后续分析的过程需要有bai文件的存在,特别是显示序列比对情况下,比如samtool的tview命令等。
#必须对bam文件进行默认情况下的排序后,才能进行index。否则会报错。
$ samtools index aln.sorted.bam(5) view:SAM<->BAM<->CRAM conversion
- 本命令将sam文件转换成bam文件;然后对bam文件进行各种操作,比如数据的排序和提取(这些操作是对bam文件进行的,不能对sam文件进行该操作);最后将排序或提取得到的数据输出为bam或sam格式。
- bam文件优点:bam文件为二进制文件,占用的磁盘空间比sam文本文件小;利用bam二进制文件的运算速度快。
#将sam文件转换成bam文件(默认情况下不加 region,则是输出所有的 region)
$ samtools view -b abc.sam > abc.bam
$ samtools view -b abc.sam -o abc.bam- 关于此部分更详细的说明请阅读 Samtools中文使用手册。
(6)depth:compute the depth
本命令对bam文件中的每个碱基位点的测序深度进行统计,并输出到标准输出。
$ samtools depth [-r reg] [-q baseQthres] [-Q mapQthres] [-b in.bed] [...] (7)mpileup:multi-way pileup
本命令用于对bam文件进行处理,生成mpileup, VCF或BCF文件,再使用bcftools或varscan2进行SNP和Indel变异位点的检测(耗时较长,且灵敏度并不高,不建议使用)。
$ samtools mpileup -gf ref.fasta [-M capMapQ] [-Q minBaseQ] [-q minMapQ] [...]= (8) fastq/a :converts a BAM to a FASTQ/A
本命令将bam文件转换为fastq或fasta格式。
$ samtools fastq [options...]