GEO数据挖掘-从数据下载到富集分析-各种绘图(学习笔记)
###############################
##---------GEO-data1---------##
###############################
rm(list = ls())
source("http://bioconductor.org/biocLite.R") #before install package
biocLite('DOSE') #bioconductor软件安装示例
## 1.下载GSE数据,获得表达量矩阵
{
suppressPackageStartupMessages(library(GEOquery))
gset <- getGEO('GSE42872',destdir = ".",
AnnotGPL = F,
getGPL = F)#下载GSE数据
save(gset,file = 'GSE42872.gset.Rdata')
exprSet <- read.table(file = 'GSE42872_series_matrix.txt.gz',
sep = '\t',
header = T,
quote = '',
fill = T,
comment.char = "!") #读取表达数据
rownames(exprSet) = exprSet[,1] #将第一列作为行名
exprSet <- exprSet[,-1] #去掉第一列
}
## 2.ID translation
View(gset) #查看对应芯片平台,“ GPL6244”,去生信菜鸟团找
suppressPackageStartupMessages(library(hugene10sttranscriptcluster.db))
ids = toTable(hugene10sttranscriptclusterSYMBOL)
length(unique(ids$symbol))
tail(sort(table(ids$symbol)))
table(sort(table(ids$symbol)))
plot(table(sort(table(ids$symbol))))
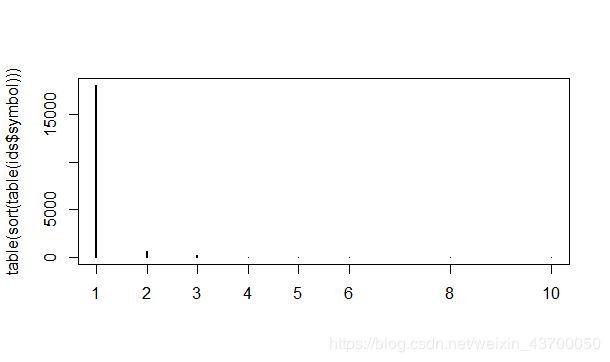
table(rownames(exprSet) %in% ids$probe_id)
exprSet = exprSet[rownames(exprSet) %in% ids$probe_id,]#初次筛选芯片
head(ids)
head(exprSet)
#exprSet[ids[,2] == 'IGKC',] #一个基因有10个芯片,需要再次过滤
#x = exprSet[ids[,2] == 'IGKC',]
#which.max(rowMeans(x))#这10个芯片在所有样本中都有均值,取均值最大的芯片
tmp = by(exprSet,
ids$symbol,
function(x) rownames(x)[which.max(rowMeans(x))])
probes = as.character(tmp)
exprSet = exprSet[rownames(exprSet) %in% probes,] #迭代,再次过滤
ids = ids[match(rownames(exprSet),ids$probe_id),]
rownames(exprSet) <- ids$symbol
exprSet1 = exprSet
save(exprSet1,file = 'exprSet_GSE42872_id_trans.Rdata')
## 3.表达矩阵的了解
library(reshape2)
library('ggplot2')
b = gset[[1]]
tmp = pData(b)
group_list = c(rep('control',3),rep('case',3))
exprSet_L = melt(exprSet1)
colnames(exprSet_L) = c('sample','value')
exprSet_L$group = rep(group_list,each = nrow(exprSet1))
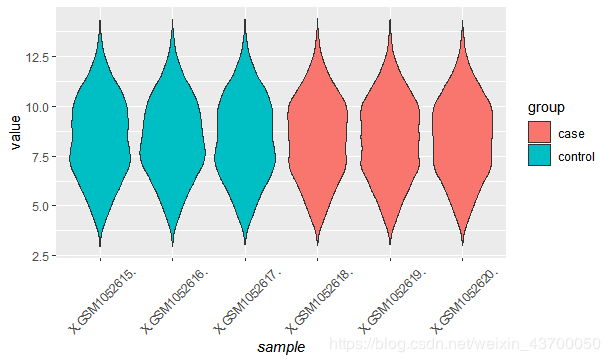
##小提琴图
p1 = ggplot(exprSet_L,aes(x = sample, y = value,fill = group)) +geom_violin()
print(p1)
##箱线图
p2 = ggplot(exprSet_L,aes(x = sample, y = value,fill = group)) +geom_boxplot()+
theme(axis.title.x = element_text(face = 'italic'),
axis.text.x = element_text(angle = 45 , vjust = 0.5))
print(p2)

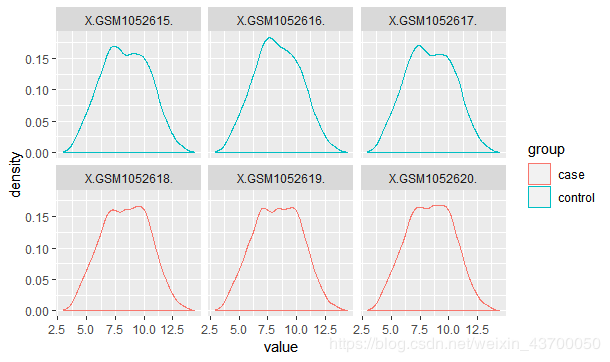
##密度图
p3 = ggplot(exprSet_L,aes(value,col=group)) +geom_density() +
facet_wrap(~sample,nrow = 2)
print(p3)
##hclust
colnames(exprSet1) = paste(group_list,1:6,sep = '') ##样本名称再次改变
nodepar = list(lab.cex = 0.6, pch = c(NA,19),
cex = 0.7,col = 'blue')
hc=hclust(dist(t(exprSet1)))
par(mar=c(5,5,5,10))
png(filename = 'hclust.png')
plot(as.dendrogram(hc),nodepar = nodepar, hariz = T)
dev.off()

##主成分分析
library(ggfortify)
df = as.data.frame(t(exprSet1))
df$group = group_list
p5 = autoplot(prcomp(df[,1:(ncol(df)-1)]),
data = df,colour = 'group')
print(p5)

## 4.差异表达
library(limma)
group_list
design <- model.matrix(~0+factor(group_list))
colnames(design) = levels(factor(group_list))
rownames(design) = colnames(exprSet1)
contrast.matrix <- makeContrasts(paste0(unique(group_list),collapse = '-'),
levels =design)
contrast.matrix[1,1] = 1 #control和case设置反了
contrast.matrix[2,1] = -1
##step1
fit <- lmFit(exprSet1,design)
##step2
fit2 <- contrasts.fit(fit,contrast.matrix)
fit2 <- eBayes(fit2)
##step3
tempOutput <- topTable(fit2, coef =1, n = Inf)
nrDEG = na.omit(tempOutput)
head(nrDEG)
save(nrDEG,file = 'nrDEG.Rdata')
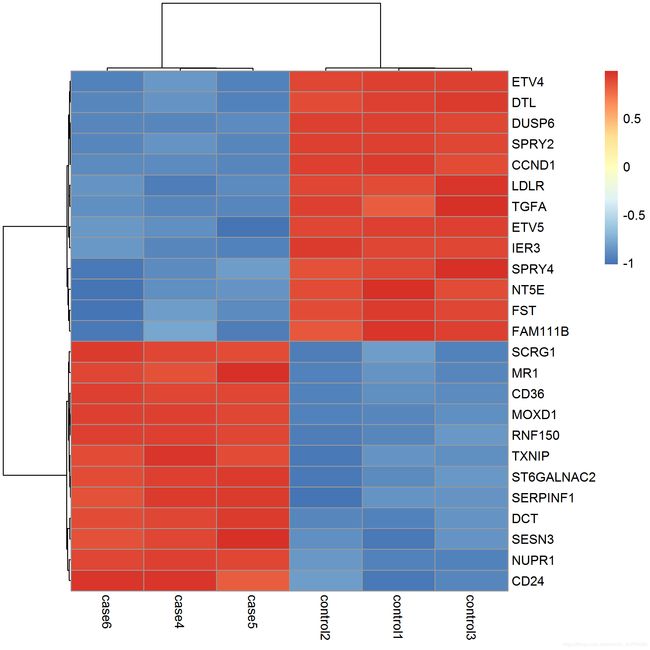
library(pheatmap)
choose_gene = head(rownames(nrDEG),25)
choose_matrix = exprSet1[choose_gene,]
choose_matrix = t(scale(t(choose_matrix)))
p = pheatmap(choose_matrix,filename = 'DEG_top25_heatmap.png')
## 5.火山图
logFC_cutoff = with(nrDEG,mean(abs(logFC))+2*sd(abs(logFC)))
nrDEG$change = as.factor(ifelse(nrDEG$P.Value < 0.05 &
abs(nrDEG$logFC) > logFC_cutoff,
ifelse(nrDEG$logFC > logFC_cutoff,
'UP','DOWN'),'NOT'))
this_title = paste0('Cutoff for logFC is ',round(logFC_cutoff,2),
'\nThe number of up gene is ',nrow(nrDEG[nrDEG$change == 'UP',]),
'\nThe number of down gene is ',nrow(nrDEG[nrDEG$change == 'DOWN',]))
library(ggplot2)
g = ggplot(data = nrDEG,
aes(x = logFC, y = -log10(P.Value),
color = change)) +
geom_point(alpha = 0.4, size = 1.75) +
theme_set(theme_set(theme_bw(base_size = 10))) +
xlab("log2 fold change") + ylab('-log10 p-value') +
ggtitle(this_title) +theme(plot.title = element_text(size = 10,hjust = 0.5)) +
scale_color_manual(values = c('blue','black','red'))
print(g)
ggsave(g,filename = 'volcano.png')

## 6.富集分析 GO KEGG DO
library(topGO)
library(Rgraphviz)
library(pathview)
library(clusterProfiler)
library(org.Hs.eg.db)
library(DOSE)
gene = rownames(nrDEG[nrDEG$P.Value < 0.05 & abs(nrDEG$logFC) > logFC_cutoff,])
gene.df <- bitr(gene, fromType = 'SYMBOL',
toType = c('ENSEMBL','ENTREZID'),
OrgDb = org.Hs.eg.db)
head(gene.df)
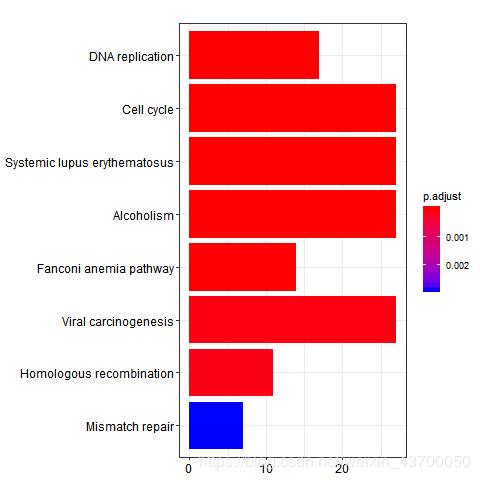
##6.1 KEGG pathway analysis
kk <- enrichKEGG(gene = gene.df$ENTREZID,
organism = 'hsa',
pvalueCutoff = 0.05)
png(filename ='kegg_barplot.png' )
barplot(kk)
dev.off()
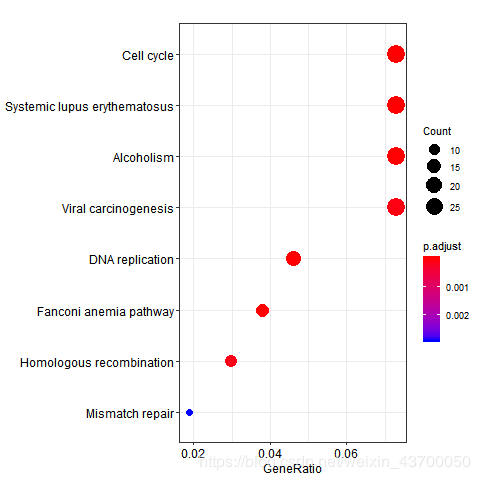
png(filename ='kegg_dotplot.png' )
dotplot(kk)
dev.off()
##6.2 GO analysis
columns(org.Hs.eg.db) #查看基因类型
enrich.go.BP = enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = 'ENTREZID',
ont = 'BP',
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = T)
png(filename ='go_bp_barplot.png' )
barplot(enrich.go.BP)
dev.off()
png(filename ='go_bp_dotplot.png' )
dotplot(enrich.go.BP)
dev.off()
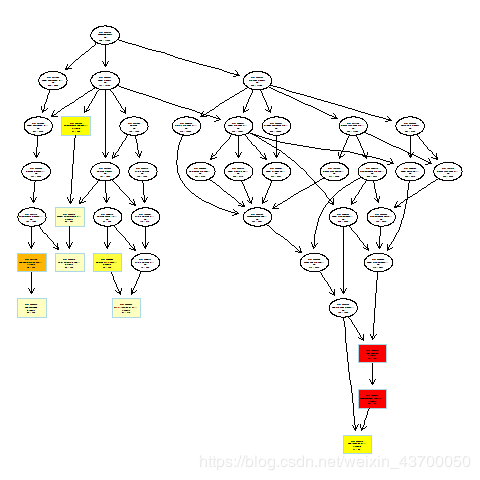
png(filename ='go_bp_plotGOgraph.png' )
plotGOgraph(enrich.go.BP)
dev.off()


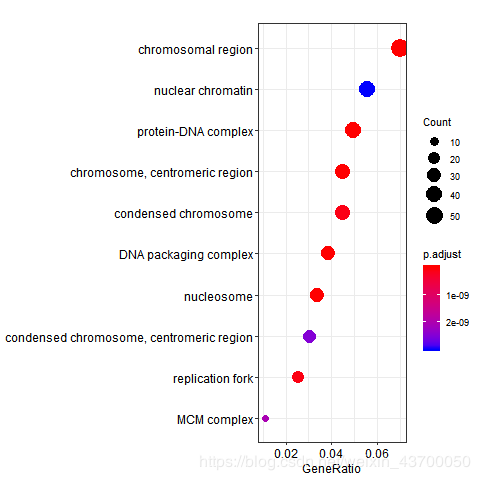
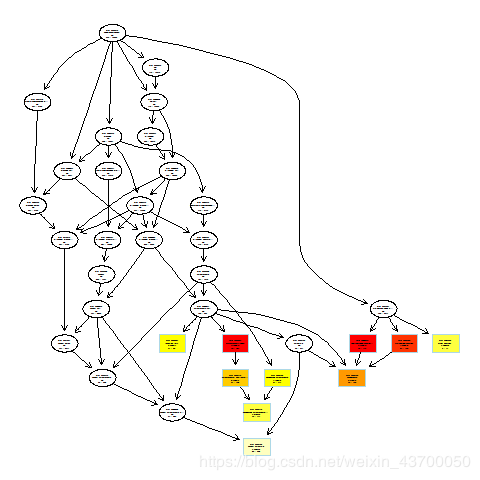
enrich.go.CC = enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = 'ENTREZID',
ont = 'CC',
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = T)
png(filename ='go_cc_barplot.png' )
barplot(enrich.go.CC)
dev.off()
png(filename ='go_cc_dotplot.png' )
dotplot(enrich.go.CC)
dev.off()
png(filename ='go_cc_plotGOgraph.png' )
plotGOgraph(enrich.go.CC)
dev.off()

enrich.go.MF = enrichGO(gene = gene.df$ENTREZID,
OrgDb = org.Hs.eg.db,
keyType = 'ENTREZID',
ont = 'MF',
pvalueCutoff = 0.01,
qvalueCutoff = 0.05,
readable = T)
png(filename ='go_mf_barplot.png' )
barplot(enrich.go.MF)
dev.off()
png(filename ='go_mf_dotplot.png' )
dotplot(enrich.go.MF)
dev.off()
png(filename ='go_mf_plotGOgraph.png' )
plotGOgraph(enrich.go.MF)
dev.off()
pdf(file ='go_mf_plotGOgraph.pdf' )
plotGOgraph(enrich.go.MF)
dev.off()



##6.3 DO analysis
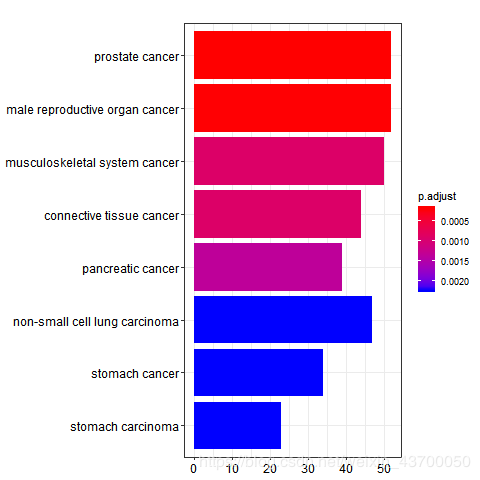
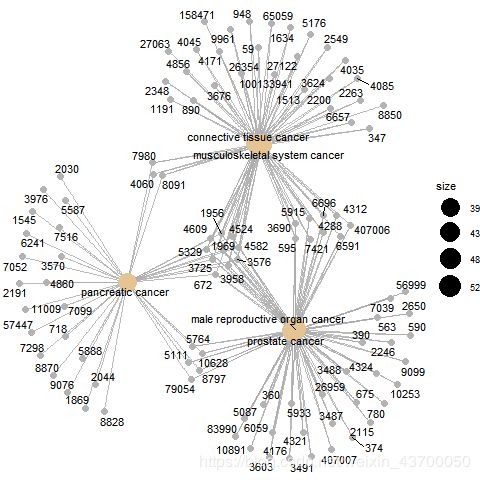
enrich.do <- enrichDO(gene = gene.df$ENTREZID,
ont = 'DO',
pvalueCutoff = 0.05,
pAdjustMethod = 'BH',
minGSSize = 5,
maxGSSize = 500,
qvalueCutoff = 0.05,
readable = F)
png(filename ='do_barplot.png' )
barplot(enrich.do)
dev.off()
png(filename ='do_dotplot.png' )
dotplot(enrich.do)
dev.off()
pdf(file ='do_cnetplot.pdf' )
cnetplot(enrich.do)
dev.off()

本博客内容将同步更新到个人微信公众号:生信玩家。欢迎大家关注~~~