更新:20190319
使用EasyCodeML完成dnds的计算,自己尝试了分支模型和位点模型的计算,简直不要太容易!
更新:20190314 教程 http://wiki.cathdb.info/wiki-beta/doku.php?id=tutorials:eccb_t2_codeml
- The selevtive presure in protein coding genes can be detected within the framework of comparative genomics.
- The selective presure is assumed to be defined by the ratio (w) dN/dS.
- dS represents the synonymous rate (changing the amino acid)
- dN represents the non-synonymous rate (keeping the amino acid)
- Under purifying selection, natural selection prevents the replacement of amino acids, so the dN will be lower than the dS.
- Values of w<1, =1, and > 1 means negative purifying selection, neutral evolution, and positive selection.
更新20190223:重复b站教学视频中dnds的计算过程
(这个视频教程不是我的,我也只是跟着这个教程来学习的!)
以下内容全部在window系统操作
使用到的软件
- MEGA 序列比对;进化树构建
下载地址 https://megasoftware.net/ - EasyCodeML 比对格式转化为PAML要求的格式
下载地址 https://github.com/BioEasy/EasyCodeML - PAML 运用分支模型估计dn/ds
下载地址 http://abacus.gene.ucl.ac.uk/software/paml.html (win系统不用编译,下载解压出来直接可以使用)
第一步:下载序列
根据教程中提到的物种拉丁名和myoglboin为关键词在NCBI中检索获得本次分析中使用到的8条肌红蛋白序列,放到同一个文件中,手动去掉终止密码子(有几条序列中不包括终止密码子,不知道为什么)(需要原始序列的同学可以给我留言)
第二步:codeML输入文件的准备
包括:序列比对;进化树构建,控制文件编辑等
- 使用MEGA进行多序列比对
读入数据,选择Alignment——Align by Muscle (Codons)
将比对结果导出为fasta格式 - 构建NJ树
读入上一步比对好的数据,构建NJ树:bootstrap1000,kumra两参数模型
 image.png
image.png
生成的结果树
 tree.PNG
tree.PNG
将生成的树文件导出为newick格式 - 使用EasyCodeml将比对结果转化为PAML要求的格式
Tools——Seqformat convertor
将上一步比对好的fasta文件拖入,第二个框输出格式选择PAML即可获得结果
参考EasyCodeML帮助文档对进化树分支进行标记 - 按照视频中的内容编辑控制文件
第三步:计算dnds
win+R快捷键输入cmd调出命令行窗口;cd命令切换到保存输出结果的文件夹下,分布输入codeml和控制文件所在的路径,回车(win下也可以拖拽)
- 零假设 所有支系共有一个omage值
控制文件内容
seqfile = C:\Users\mingy\Desktop\practice\dnds\myoglobin.pml * sequence data filename
treefile = C:\Users\mingy\Desktop\practice\dnds\myoglobin_tree.nwk * tree structure file name
outfile = C:\Users\mingy\Desktop\practice\dnds\result_null.txt * main result file name
noisy = 9 * 0,1,2,3,9: how much rubbish on the screen
verbose = 1 * 0: concise; 1: detailed, 2: too much
runmode = 0 * 0: user tree; 1: semi-automatic; 2: automatic
* 3: StepwiseAddition; (4,5):PerturbationNNI; -2: pairwise
seqtype = 1 * 1:codons; 2:AAs; 3:codons-->AAs
CodonFreq = 2 * 0:1/61 each, 1:F1X4, 2:F3X4, 3:codon table
* ndata = 10
clock = 0 * 0:no clock, 1:clock; 2:local clock; 3:CombinedAnalysis
aaDist = 0 * 0:equal, +:geometric; -:linear, 1-6:G1974,Miyata,c,p,v,a
aaRatefile = dat/jones.dat * only used for aa seqs with model=empirical(_F)
* dayhoff.dat, jones.dat, wag.dat, mtmam.dat, or your own
model = 0
* models for codons:
* 0:one, 1:b, 2:2 or more dN/dS ratios for branches
* models for AAs or codon-translated AAs:
* 0:poisson, 1:proportional, 2:Empirical, 3:Empirical+F
* 6:FromCodon, 7:AAClasses, 8:REVaa_0, 9:REVaa(nr=189)
NSsites = 0 * 0:one w;1:neutral;2:selection; 3:discrete;4:freqs;
* 5:gamma;6:2gamma;7:beta;8:beta&w;9:betaγ
* 10:beta&gamma+1; 11:beta&normal>1; 12:0&2normal>1;
* 13:3normal>0
icode = 0 * 0:universal code; 1:mammalian mt; 2-10:see below
Mgene = 0
* codon: 0:rates, 1:separate; 2:diff pi, 3:diff kapa, 4:all diff
* AA: 0:rates, 1:separate
fix_kappa = 0 * 1: kappa fixed, 0: kappa to be estimated
kappa = 2 * initial or fixed kappa
fix_omega = 0 * 1: omega or omega_1 fixed, 0: estimate
omega = .4 * initial or fixed omega, for codons or codon-based AAs
fix_alpha = 1 * 0: estimate gamma shape parameter; 1: fix it at alpha
alpha = 0. * initial or fixed alpha, 0:infinity (constant rate)
Malpha = 0 * different alphas for genes
ncatG = 8 * # of categories in dG of NSsites models
getSE = 0 * 0: don't want them, 1: want S.E.s of estimates
RateAncestor = 1 * (0,1,2): rates (alpha>0) or ancestral states (1 or 2)
Small_Diff = .5e-6
cleandata = 1 * remove sites with ambiguity data (1:yes, 0:no)?
* fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed, 3: proportional
method = 0 * Optimization method 0: simultaneous; 1: one branch a time
* Genetic codes: 0:universal, 1:mammalian mt., 2:yeast mt., 3:mold mt.,
* 4: invertebrate mt., 5: ciliate nuclear, 6: echinoderm mt.,
* 7: euplotid mt., 8: alternative yeast nu. 9: ascidian mt.,
* 10: blepharisma nu.
* These codes correspond to transl_table 1 to 11 of GENEBANK.
这里的参数很多都不明白,前几天大体看了一下paml的文档,里面有很详细的介绍,需要花时间多看
关键输出结果
lnL(ntime: 13 np: 15): -1284.006906 +0.000000
kappa (ts/tv) = 1.86874
omega (dN/dS) = 0.13353
dN & dS for each branch
branch t N S dN/dS dN dS N*dN S*dS
9..10 0.050 382.1 79.9 0.1335 0.0079 0.0588 3.0 4.7
10..11 0.108 382.1 79.9 0.1335 0.0170 0.1271 6.5 10.2
11..12 0.040 382.1 79.9 0.1335 0.0062 0.0467 2.4 3.7
12..1 0.079 382.1 79.9 0.1335 0.0124 0.0932 4.8 7.4
12..6 0.106 382.1 79.9 0.1335 0.0166 0.1242 6.3 9.9
11..13 0.073 382.1 79.9 0.1335 0.0114 0.0853 4.4 6.8
13..4 0.000 382.1 79.9 0.1335 0.0000 0.0000 0.0 0.0
13..8 0.027 382.1 79.9 0.1335 0.0042 0.0314 1.6 2.5
10..14 0.172 382.1 79.9 0.1335 0.0270 0.2024 10.3 16.2
14..2 0.052 382.1 79.9 0.1335 0.0081 0.0608 3.1 4.9
14..5 0.037 382.1 79.9 0.1335 0.0059 0.0440 2.2 3.5
9..7 0.116 382.1 79.9 0.1335 0.0182 0.1361 6.9 10.9
9..3 0.113 382.1 79.9 0.1335 0.0177 0.1326 6.8 10.6
tree length for dN: 0.1526
tree length for dS: 1.1425
这里的输出结果和视频教程中的略有差别,视频中的ntime和np分别是14和16,暂时还没有搞懂问题出在哪里
- 备择假设:特定支系拥有不同的omega值
只需要将树文件替换为标记分支的树文件,model值改为2即可
lnL(ntime: 13 np: 16): -1282.758923 +0.000000
kappa (ts/tv) = 1.86014
w (dN/dS) for branches: 0.11905 0.37136
dN & dS for each branch
branch t N S dN/dS dN dS N*dN S*dS
9..10 0.053 382.2 79.8 0.1190 0.0077 0.0647 2.9 5.2
10..11 0.095 382.2 79.8 0.3714 0.0245 0.0661 9.4 5.3
11..12 0.040 382.2 79.8 0.1190 0.0058 0.0486 2.2 3.9
12..1 0.081 382.2 79.8 0.1190 0.0118 0.0990 4.5 7.9
12..6 0.105 382.2 79.8 0.1190 0.0154 0.1290 5.9 10.3
11..13 0.073 382.2 79.8 0.1190 0.0106 0.0893 4.1 7.1
13..4 0.000 382.2 79.8 0.1190 0.0000 0.0000 0.0 0.0
13..8 0.027 382.2 79.8 0.1190 0.0039 0.0328 1.5 2.6
10..14 0.177 382.2 79.8 0.1190 0.0259 0.2176 9.9 17.4
14..2 0.054 382.2 79.8 0.1190 0.0078 0.0658 3.0 5.3
14..5 0.036 382.2 79.8 0.1190 0.0052 0.0439 2.0 3.5
9..7 0.117 382.2 79.8 0.1190 0.0171 0.1434 6.5 11.5
9..3 0.114 382.2 79.8 0.1190 0.0167 0.1406 6.4 11.2
tree length for dN: 0.1525
tree length for dS: 1.1409
lnl_null - lnl_alternative 符合卡方分布
使用R语言的phisq函数计算显著性
自己得到的结果是1,没搞清楚问题出在哪里
步骤应该就是这么个步骤,关于结果的解读和其他模型的设置多看paml的帮助手册
1、下载安装
第一种方法
下载地址 http://abacus.gene.ucl.ac.uk/software/paml4.9h.tgz
安装教程 http://abacus.gene.ucl.ac.uk/software/paml.html
第二种方法
直接用conda,conda的安装方法可以参考公众号 生信技能树 的文章价值999的全外显子教学视频--免费送
conda安装好以后直接使用命令
conda install paml
2、准备分析dnds比值所需要的文件
将CDS序列进行基于密码子的比对,这一步可借助mega完成,然后导出比对结果为fasta格式,然后按照视频教程去掉终止密码子并整理成所需要的格式(所需要的格式第一行是tab+数字+tab+数字,第一个数字是几条序列,第二个数字是每条序列有几个位点,自己获得以上信息的做法是将fasta格式的比对文件转换成phylip格式,可以实现比对文件格式转换的小工具http://sing.ei.uvigo.es/ALTER/);树文件可以用IQ-tree获得,IQ-tree下载及安装http://www.iqtree.org/#download
3、实例:计算五种植物叶绿体的ccsA基因的dn/ds值
A、比对:打开mega, File——Open a File 选择文件,提示


这里需要注意的地方是序列名后有两个空格
B、建树:IQ-tree解压出来后bin目录下有可执行的程序iqtree,运行
./iqtree
建树用到的命令是
./iqtree -s alignment.file -alrt 1000 -bb 1000
C、按照视频教程的内容修改控制文件的内容,然后执行程序即可。获得的结果
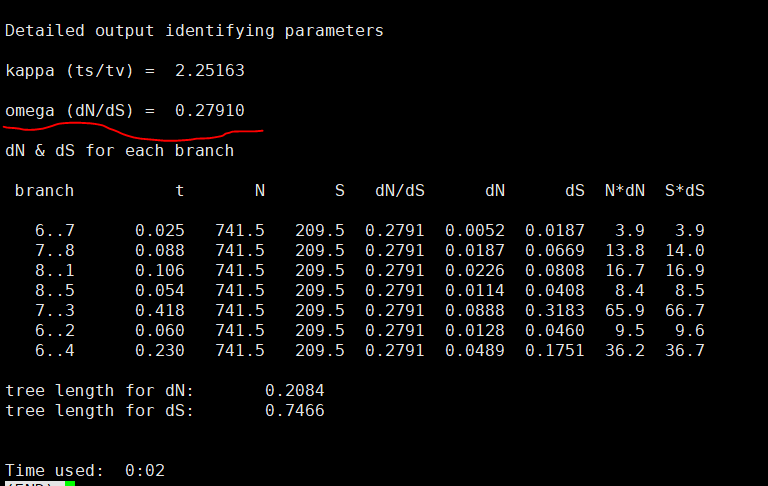
以上分析用到的CDS序列文件 https://pan.baidu.com/s/1xyNj_cvR68An-Lkf2HGBgQ
发现一个小问题:如果使用conda install paml 安装paml 好像找不到需要的控制文件在哪
以上内容基本在linux系统操作完成,如果没有linux系统,目前想到三种解决办法:
1、如果自己电脑是win10系统可以直接安装linux系统,百度搜索相关教程即可;
2、购买腾讯云,学生套餐10块每月还挺划算的,1核2G运行内存,50G存储空间;
3、安装虚拟机,自己之前也尝试过,个人感觉稍微有些麻烦。
关于linux系统的入门学习个人强推 网易云课堂 细说linux 视频教程
更新
配置文件的参数修改
前三个参数用来指定文件位置
runmode = 0 user tree
seqtype = 1 codons
ndata = 自己暂时还不知道这个参数是用来做什么的,视频教程里的配置文件没有这个参数
model = 0
Reference
https://www.plob.org/article/6943.html codeml的配置文件
https://www.bilibili.com/video/av10469605?from=search&seid=8859495264060497812 b站教学视频
微信文章: Ka/Ks比值:核酸分子进化的指标
微信文章: 从人见人爱的向日葵说起——Ks与全基因组多倍化事件
微信文章: 价值999的全外显子教学视频--免费送
微信文章: 基于ML法重建系统发育树-IQ-TREE篇
http://www.biotrainee.com/thread-1685-1-1.html 生信技能树
https://wenku.baidu.com/view/39c6d6f30029bd64783e2c60.html 百度文库