本文首发于我的个人博客, http://xuzhougeng.top/
往期回顾:
- 使用ArchR分析单细胞ATAC-seq数据(第一章)
- 使用ArchR分析单细胞ATAC-seq数据(第二章)
- 使用ArchR分析单细胞ATAC-seq数据(第三章)
- 使用ArchR分析单细胞ATAC-seq数据(第四章)
- 使用ArchR分析单细胞ATAC-seq数据(第五章)
- 使用ArchR分析单细胞ATAC-seq数据(第六章)
- 使用ArchR分析单细胞ATAC-seq数据(第七章)
- 使用ArchR分析单细胞ATAC-seq数据(第八章)
- 使用ArchR分析单细胞ATAC-seq数据(第九章)
- 使用ArchR分析单细胞ATAC-seq数据(第十章)
- 使用ArchR分析单细胞ATAC-seq数据(第十一章)
- 使用ArchR分析单细胞ATAC-seq数据(第十二章)
- 使用ArchR分析单细胞ATAC-seq数据(第十三章)
- 使用ArchR分析单细胞ATAC-seq数据(第十四章)
第15章 使用ArchR进行整合分析
ArchR的一个优势能够整合多种水平的信息从而提供新的洞见。我们可以只用ATAC-seq数据进行分析,如识别peak之间的共开放性来预测调控相互作用,或整合scRNA-seq数据,如通过peak-基因的连锁分析预测增性子活性。无论是哪种情况,ArchR都可以很容易地从scATAC seq数据中获得更深入的见解。
15.1 创建细胞低重叠聚集
ArchR能方便许多特征间相关性的分析。在这些相关分析中,使用稀疏的单细胞数据进行这些计算会导致大量的噪声。为规避这一挑战,我们采用了一种由Cicero引入的方法,在这些分析之前创建单细胞的低重叠聚集。我们过滤与任何其他聚集重叠超过80%的聚集以减少偏差。同时为提升该方法的速度,我们开发了一个优化的迭代重叠检查流程,借由"Rcpp"包通过C++实现了快速特征相关性运算。在ArchR中,这些优化方法被用于计算peak共开放性、peak-基因连锁和其他连锁分析。这些低重叠聚集运算都是在内部完成,为清楚起见,我们先在这里对其介绍。
15.2 ArchR共开放分析
共开放是多个单细胞种的两个peak之间开放性的相关性。换句话说,当peak A在某个单细胞中是开放状态时,peak B通常也是开放状态。下图可以直观地说明了这一概念,说明增强子E3通常与启动子P是共开放的。
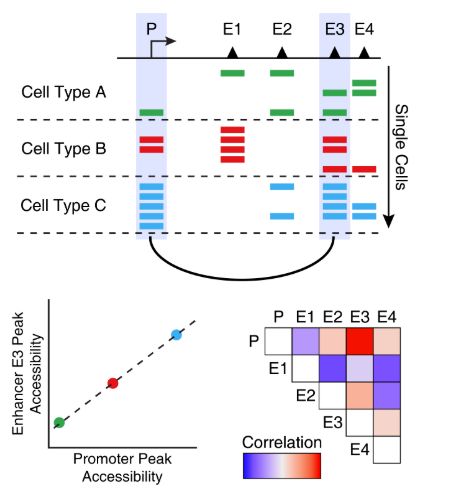
有一点需要注意,共开放分析找到的peak通常都是细胞类型特异的peak。这是因为这些peak在一种细胞类型中都是开放的,而另一种细胞类型通常都是关闭的。虽然这些peak之间有很强的相关性,但是不意味着这些peak之间存在调控关系。
在ArchR中,我们使用addCoAccessibility()函数计算共开放性,计算结束得到的开放性信息保存在ArchRProject中
projHeme5 <- addCoAccessibility(
ArchRProj = projHeme5,
reducedDims = "IterativeLSI"
)使用getCoAccessibility()函数可以从ArchRProject对象中提取共开放性信息,当设置returnLoops=FALSE时会返回一个DataFrame对象。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1,
returnLoops = FALSE
)该DataFrame对象包含几个重要的信息。queryHits和subjectHits列记录的是两个存在相关性的peak的索引。correlation则是两个peak之间的开放状态相关的数值。
cA
# DataFrame with 64824 rows and 4 columns
# queryHits subjectHits seqnames correlation
#
# 1 5 10 chr1 0.63855416236086
# … … … … …
# 64823 143977 143978 chrX 0.550319862774772这个共开放DataFrame还有一个元数据成员,包含一个相关peak的GRanges对象。上面提到的queryHits和subjectHits的索引适用于这个GRanges对象。
metadata(cA)[[1]]
# GRanges object with 144009 ranges and 0 metadata columns:
# seqnames ranges strand
#
# Mono chr1 752499-752999 *
# NK chr1 762651-763151 *
# B chr1 801006-801506 *
# B chr1 805039-805539 *
# CLP chr1 845325-845825 *
# … … … …
# Erythroid chrX 154664540-154665040 *
# NK chrX 154807324-154807824 *
# PreB chrX 154840785-154841285 *
# PreB chrX 154842404-154842904 *
# NK chrX 154862017-154862517 *
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths如果我们设置retureLoops=TRUE, 那么getCoAccessibility()会以loop track的形式返回共开放数据。在这个GRanges对象中,IRanges的起始和结束对应的是每个存在互作关系中两个共开放peak的位置。resolution参数设置这些loop的碱基对分辨率。当resolution=1时,它会输出连接每个peak中心的loop.
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1,
returnLoops = TRUE
)我们比较下GRanges和DataFrame这两个对象
cA[[1]]
# GRanges object with 32412 ranges and 1 metadata column:
# seqnames ranges strand | value
# [1] chr1 845575-856640 * | 0.63855416236086
# … … … … . …
# [32412] chrX 153980218-153990364 * | 0.550319862774772
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths如果我们将loops的分辨率调整为resolution = 1000, 这能够避免绘制过多的共开放互作事件。从下面的输出的GRanges对象中,我们可以看到条目少了很多
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 1000,
returnLoops = TRUE
)
cA[[1]]
# GRanges object with 30997 ranges and 1 metadata column:
# seqnames ranges strand | value
# |
# [1] chr1 845500-856500 * | 0.63855416236086
# … … … … . …
# [30997] chrX 153980500-153990500 * | 0.550319862774772
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths同样的,如果我们进一步降低分辨率resolution = 10000,我们会得到更少共开放交互事件。
cA <- getCoAccessibility(
ArchRProj = projHeme5,
corCutOff = 0.5,
resolution = 10000,
returnLoops = TRUE
)
cA[[1]]
# GRanges object with 21142 ranges and 1 metadata column:
# seqnames ranges strand | value
# |
# [1] chr1 845000-855000 * | 0.63855416236086
# … … … … . …
# [21142] chrX 153985000-153995000 * | 0.550319862774772
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths15.2.1 在browser track中绘制共开放
当我们在ArchRProject里增加共开放信息后,我们可以用这个信息在基因组浏览器里绘制loop track。具体做法就是在plotBrowserTrack()函数中设置loops参数。我们这里以getCoAccessibility()默认参数输出结果为例,即corCutOff = 0.5, resolution = 1000, returnLoops = TRUE
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p <- plotBrowserTrack(
ArchRProj = projHeme5,
groupBy = "Clusters2",
geneSymbol = markerGenes,
upstream = 50000,
downstream = 50000,
loops = getCoAccessibility(projHeme5)
)最后用grid.draw函数绘制结果,通过$选择特定的标记基因。
grid::grid.newpage()
grid::grid.draw(p$CD14)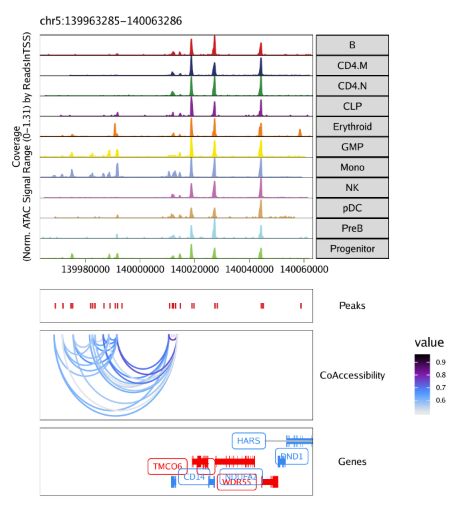
使用plotPDF()保存可编辑的矢量图
plotPDF(plotList = p,
name = "Plot-Tracks-Marker-Genes-with-CoAccessibility.pdf",
ArchRProj = projHeme5,
addDOC = FALSE, width = 5, height = 5)15.3 ArchR Peak2GeneLinkages分析
和共开放分析类似,ArchR也能分析所谓的"peak-to-gene"关联, 也就是分析peak和基因的相关性。共开放分析和peak-to-gene关联分析的主要不同在于,共开放分析只需要用到ATAC-seq数据,寻找的是peak之间的共开放关系,而peak-to-gene关联分析则会整合scRNA-seq数据,寻找peak开放状态和基因表达量的关系。这其实是相似问题的两种方法。只不过因为peak-to-gene关联分析使用的是scATAC-seq和scRNA-seq数据,我们会认为这个关联更能反应基因调控关系。
在ArchR中,我们使用addPeak2GeneLinks()函数鉴定peak-to-gene的关联。
projHeme5 <- addPeak2GeneLinks(
ArchRProj = projHeme5,
reducedDims = "IterativeLSI"
)我们以上一节类似方式提取这些peak-to-gene的关联信息,只不过这里用的是getPeak2GeneLinks()函数。和之前一样,用户可以设置相关性阈值和关联之间的分辨率。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1,
returnLoops = FALSE
)当returnLoops=FALSE, 该函数返回一个DataFrame对象,和之前getCoAccessibility()返回的DataFrame相似。主要的不同在于scATAC-seq的索引 peak存放在idxATAC列,而scRNA-seq基因的索引是存放在idxRNA列中
p2g
# DataFrame with 43754 rows and 6 columns
# idxATAC idxRNA Correlation FDR
#
# 1 47 5 0.549552663393716 1.34094093110629e-38
# 2 3 6 0.487418258348982 2.1460798658766e-29
...
# VarQATAC VarQRNA
#
# 1 0.948753202924817 0.793290683296597
# 2 0.253206396822421 0.445890005913661peak-to-gene关联DataFrame对象同样也有一个GRanges对象记录对应peak的元信息。上面提到的idxATAC索引能用于GRanges对象中。
metadata(p2g)[[1]]
# GRanges object with 144009 ranges and 0 metadata columns: ## seqnames ranges strand
#
# [1] chr1 752499-752999 *
# … … … …
# [144009] chrX 154862017-154862517 *
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths如果们设置returnLoop=TRUE,那么getPeak2GeneLinks()会返回一个记录loop track的GRanges对象,链接peak和基因。和之前共开放分析一样,IRanges对象里记录着关联着的peak和基因的位置。当resolution=1, 这连接的是peak的中心和基因的单碱基TSS。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1,
returnLoops = TRUE
)
p2g[[1]]
# GRanges object with 43695 ranges and 2 metadata columns: ## seqnames ranges strand | value
# |
# [1] chr1 762901-948847 * | 0.533350285896763
# … … … … . …
# [43695] chrX 154444701-154664790 * | 0.493389088498317
# FDR
#
# [1] 5.20045729958651e-36
# … …
# [43695] 3.37643947034054e-30
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths我们可以将loops的分辨率调整为resolution = 1000, 输出结果主要用于在基因组浏览器中绘制这些连接。因为相同的基因对应的peak过多时,可视化效果会很差。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 1000,
returnLoops = TRUE
)
p2g[[1]]
# GRanges object with 42126 ranges and 2 metadata columns:
# seqnames ranges strand | value
# |
# [1] chr1 762500-948500 * | 0.533350285896763
# … … … … . …
# [42126] chrX 154444500-154664500 * | 0.493389088498317
# FDR
#
# [1] 5.20045729958651e-36
# … …
# [42126] 3.37643947034054e-30
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths进一步降低分辨率,也会降低peak-to-gene连接的总数。
p2g <- getPeak2GeneLinks(
ArchRProj = projHeme5,
corCutOff = 0.45,
resolution = 10000,
returnLoops = TRUE
)
p2g[[1]]
# GRanges object with 33645 ranges and 2 metadata columns:
# seqnames ranges strand | value
# |
# [1] chr1 765000-945000 * | 0.533350285896763
# … … … … . …
# [33645] chrX 154445000-154665000 * | 0.493389088498317
# FDR
#
# [1] 5.20045729958651e-36
# … …
# [33645] 3.37643947034054e-30
# ——-
# seqinfo: 23 sequences from an unspecified genome; no seqlengths15.3.1 在browser track中绘制peak-to-gene连接
我们可以采用之前共开放分析流程中的相同方法,使用plotBrowserTrack()函数在基因组浏览器上绘制peak-to-gene 连接。
markerGenes <- c(
"CD34", #Early Progenitor
"GATA1", #Erythroid
"PAX5", "MS4A1", #B-Cell Trajectory
"CD14", #Monocytes
"CD3D", "CD8A", "TBX21", "IL7R" #TCells
)
p <- plotBrowserTrack(
ArchRProj = projHeme5,
groupBy = "Clusters2",
geneSymbol = markerGenes,
upstream = 50000,
downstream = 50000,
loops = getPeak2GeneLinks(projHeme5)
)用grid.draw函数绘制结果,通过$选择特定的标记基因。
grid::grid.newpage()
grid::grid.draw(p$CD14)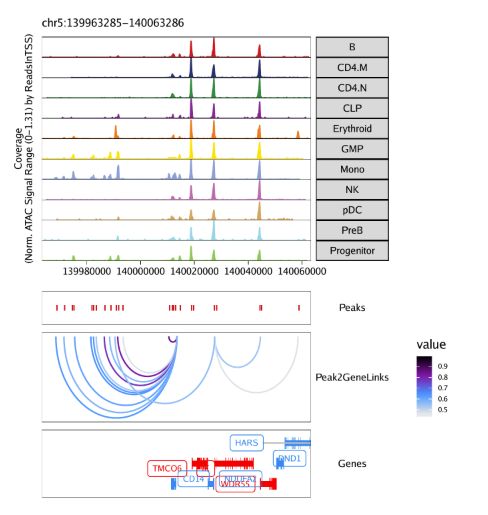
使用plotPDF()保存可编辑的矢量图
plotPDF(plotList = p,
name = "Plot-Tracks-Marker-Genes-with-Peak2GeneLinks.pdf",
ArchRProj = projHeme5,
addDOC = FALSE, width = 5, height = 5)15.3.2 绘制Peak-to-gene连接热图
我们还可以通过绘制peak-to-gene热图的方式展示我们的peak-to-gene连接。热图分为两个部分,一个是scATAC-seq数据,一个是scRNA-seq数据。绘图使用plotPeak2GeneHeatmap()函数
p <- plotPeak2GeneHeatmap(ArchRProj = projHeme5, groupBy = "Clusters2")热图的行是k-means聚类结果,其中k由我们进行设置,默认是25.
p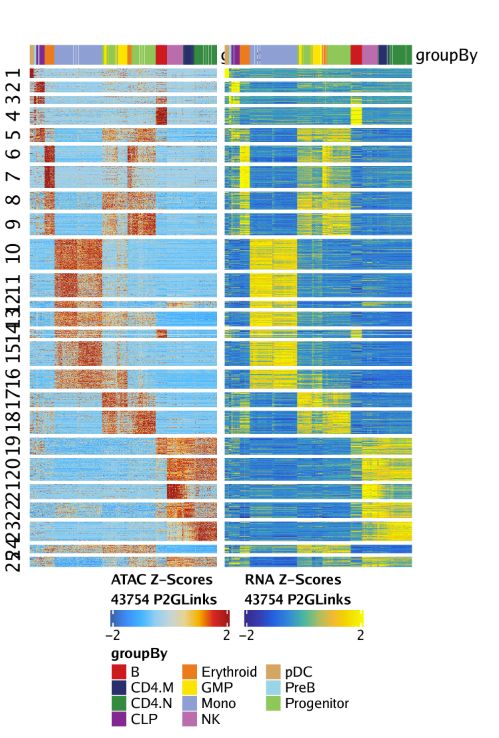
15.4 正向TF-调控因子鉴定
ATAC-seq还可以无偏好的鉴定TF,这些TF所对应的染色质区域(存在该TF结合的motif DNA序列 )表现出很大改变。此外,从聚集的位置权重矩阵(Position Weight Matrices, PWM)中也能看到这些TF家族在其集合位点上有相似的特征(例如 GATA因子)。

这种motif的相似性使得识别特定的TF充满了挑战性,这些TF可能在其预测的结合位点驱动染色质开放状态发生改变。为了规避这一挑战,我们之前用ATAC-seq和RNA-seq来鉴定TF,这些TF的基因表达和对应motif的开放状态呈正相关。我们将这些TF命名为"正向调节因子"。然而,这种分析依赖于匹配的基因表达数据,而这些数据并不是所有实验中都有。ArchR可以识别其推断基因得分与其chromVAR TF偏差z-score相关的TF来克服这种依赖性。实现方式为,ArchR利用低重叠细胞聚集将TF motif的chromVAR deviation z-scroe和TF基因的基因活跃得分相关联。当ArchR整合scRNA-seq时,TF的基因表达可以代替推测的基因活性评分。
15.4.1 第一步: 鉴定偏离TF motif
鉴定正向TF调控因子的第一步就是鉴定存在偏离的TF motif。我们在之前的章节进行过该分析,为所有的motif创建了一个MotifMatrix,记录着chromVAR deviations 和deviation z-scores。使用getGroupSE()函数可以获取基于聚类平均后的数据,返回的是一个SummarizedExperiment.
seGroupMotif <- getGroupSE(ArchRProj = projHeme5, useMatrix = "MotifMatrix", groupBy = "Clusters2")因为SummarizedExperiment对象来自于MotifMatrix, 所以它有两个seqnames, "deviation"和"z",对应chromVAR里原始的偏离值和偏离值的z-scores.
seGroupMotif
# class: SummarizedExperiment
# dim: 1740 11
# metadata(0):
# assays(1): MotifMatrix
# rownames(1740): f1 f2 … f1739 f1740
# rowData names(3): seqnames idx name
# colnames(11): B CD4.M … PreB Progenitor
# colData names(16): TSSEnrichment ReadsInTSS … FRIP nCells我们可以从SummarizedExperiment中只提取deviation z-scores.
seZ <- seGroupMotif[rowData(seGroupMotif)$seqnames=="z",]然后,我们计算该z-score在所有聚类中最大变化值。根据motif在不同聚类中观察到的差异程度,我们可以对其分层。
rowData(seZ)$maxDelta <- lapply(seq_len(ncol(seZ)), function(x){
rowMaxs(assay(seZ) - assay(seZ)[,x])
}) %>% Reduce("cbind", .) %>% rowMaxs15.4.2 第二步: 鉴定相关的TF motif和TF基因得分/表达值
接着我们使用correlateMatrices()函数来获取motif开放状态矩阵和基因活跃矩阵(基因得分或基因表达量),在这里就是GeneScoreMatrix和MotifMatrix. 它们将用于鉴定一类motif开放状态和其自身基因活跃相关的TF。正如之前所提到,我们使用许多低重叠细胞聚集在低维空间里(对应reducedDims参数)来计算相关性。
corGSM_MM <- correlateMatrices(
ArchRProj = projHeme5,
useMatrix1 = "GeneScoreMatrix",
useMatrix2 = "MotifMatrix",
reducedDims = "IterativeLSI"
)该函数返回一个DataFrame对象,记录着GeneScoreMatrix和MotifMatrix,以及它们在低重叠细胞聚类中的相关性。
corGSM_MM
# DataFrame with 825 rows and 14 columns
# GeneScoreMatrix_name MotifMatrix_name cor
#
# 1 HES4 HES4_95 0.154455056755803
# … … … …
# 825 MECP2 MECP2_645 0.0629645419544807
# padj pval GeneScoreMatrix_seqnames
#
# 1 0.48577773775473 0.000593860315103582 chr1
# … … … …
# 825 1 0.163611488297825 chrX
# GeneScoreMatrix_start GeneScoreMatrix_end GeneScoreMatrix_strand
#
# 1 935552 934342 2
# … … … …
# 825 153363188 153287264 2
# GeneScoreMatrix_idx GeneScoreMatrix_matchName MotifMatrix_seqnames
#
# 1 15 HES4 z
# … … … …
# 825 874 MECP2 z
# MotifMatrix_idx MotifMatrix_matchName
#
# 1 95 HES4
# … … …
# 825 645 MECP2同样的分析还可以使用GeneIntegrationMatrix, 用于替代GeneScoreMatrix
corGIM_MM <- correlateMatrices(
ArchRProj = projHeme5,
useMatrix1 = "GeneIntegrationMatrix",
useMatrix2 = "MotifMatrix",
reducedDims = "IterativeLSI"
)15.4.3 第三步: 在相关性DataFrame中添加极大偏差值
对于每个相关性分析,我们使用第一步计算的极大偏差值(maximum delta )来注释每个motif
corGSM_MM$maxDelta <- rowData(seZ)[match(corGSM_MM$MotifMatrix_name, rowData(seZ)$name), "maxDelta"]
corGIM_MM$maxDelta <- rowData(seZ)[match(corGIM_MM$MotifMatrix_name, rowData(seZ)$name), "maxDelta"]15.4.4 第四步: 鉴定正向TF调控因子
我们可以利用所有这些信息来识别正向TF调控因子。在下面的例子中,正向调控因子的标准为
- motif和基因得分(或基因表达)之间的相关性大于0.5
- 调整后的p值小于0.01
- deviation z-score的最大组间差异位于前四分位。
我们应用这些过滤标准,并且做一些文本调整来分离TF名字。
corGSM_MM <- corGSM_MM[order(abs(corGSM_MM$cor), decreasing = TRUE), ]
corGSM_MM <- corGSM_MM[which(!duplicated(gsub("\\-.*","",corGSM_MM[,"MotifMatrix_name"]))), ]
corGSM_MM$TFRegulator <- "NO"
corGSM_MM$TFRegulator[which(corGSM_MM$cor > 0.5 & corGSM_MM$padj < 0.01 & corGSM_MM$maxDelta > quantile(corGSM_MM$maxDelta, 0.75))] <- "YES"
sort(corGSM_MM[corGSM_MM$TFRegulator=="YES",1])
# [1] “ATOH1” “BCL11A” “CEBPA-DT” “CEBPB” “CEBPD” “CREB1”
# [7] “CREB3L4” “EBF1” “EGR2” “EOMES” “ERF” “ESR1”
# [13] “ETS1” “ETV3” “FUBP1” “GATA1” “GATA2” “GATA5”
# [19] “GATA6” “IRF1” “JDP2” “KLF11” “KLF2” “LYL1”
# [25] “MECOM” “MITF” “NFE2” “NFIA” “NFIB” “NFIC”
# [31] “NFIX” “NHLH1” “POU2F1” “RUNX2” “SIX5” “SMAD1”
# [37] “SMAD9” “SP4” “SPI1” “SPIB” “TAL1” “TCF15”
# [43] “TCF23” “TCF4” “TFAP2C” “TWIST1” “TWIST2” “YY1”
# [49] “ZEB1-AS1”在从基因得分和motif deviation z-scores鉴定到正向TF调控因子后,我们可以在点图中突出展示
p <- ggplot(data.frame(corGSM_MM), aes(cor, maxDelta, color = TFRegulator)) +
geom_point() +
theme_ArchR() +
geom_vline(xintercept = 0, lty = "dashed") +
scale_color_manual(values = c("NO"="darkgrey", "YES"="firebrick3")) +
xlab("Correlation To Gene Score") +
ylab("Max TF Motif Delta") +
scale_y_continuous(
expand = c(0,0),
limits = c(0, max(corGSM_MM$maxDelta)*1.05)
)
p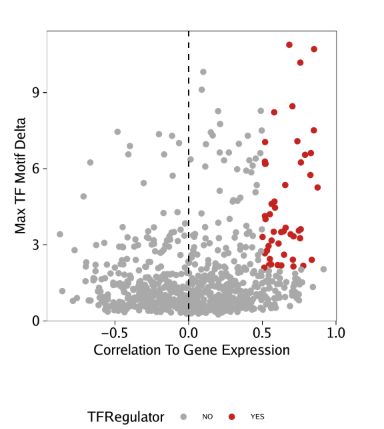
使用相同的分析策略处理GeneIntegrationMatrix
corGIM_MM <- corGIM_MM[order(abs(corGIM_MM$cor), decreasing = TRUE), ]
corGIM_MM <- corGIM_MM[which(!duplicated(gsub("\\-.*","",corGIM_MM[,"MotifMatrix_name"]))), ]
corGIM_MM$TFRegulator <- "NO"
corGIM_MM$TFRegulator[which(corGIM_MM$cor > 0.5 & corGIM_MM$padj < 0.01 & corGIM_MM$maxDelta > quantile(corGIM_MM$maxDelta, 0.75))] <- "YES"
sort(corGIM_MM[corGIM_MM$TFRegulator=="YES",1])
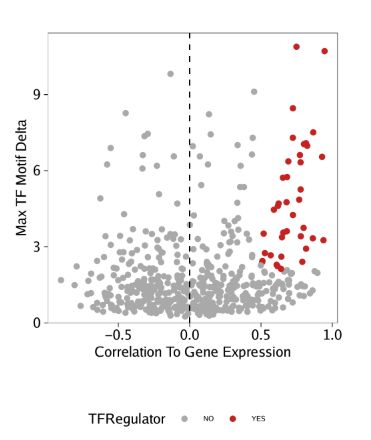
p <- ggplot(data.frame(corGIM_MM), aes(cor, maxDelta, color = TFRegulator)) +
geom_point() +
theme_ArchR() +
geom_vline(xintercept = 0, lty = "dashed") +
scale_color_manual(values = c("NO"="darkgrey", "YES"="firebrick3")) +
xlab("Correlation To Gene Expression") +
ylab("Max TF Motif Delta") +
scale_y_continuous(
expand = c(0,0),
limits = c(0, max(corGIM_MM$maxDelta)*1.05)
)
p