tags: NGS duplication
不管从哪个角度看我们都希望测序仪产出的数据中 duplicate 率尽量低。怎样降低 duplicate 率? 构建文库时,核酸提取质量要好,起始 DNA 量要足够多,建库过程中 PCR 循环数尽量少,可以的话构建 PCR-free 文库最好,防范于未然。
什么样的数据算是 duplicate
duplicate 就是一段序列的多个拷贝,以 PE 测序为例,用比对软件在将测序 reads 比对到参考基因组之后,如果有两对 reads 的 read1 和 read2 都完全比对到参考基因组上的相同位置,其中一对 reads 会被标记为 duplicate 。我画了一个示意图:

图中 A 和 B 这两对 reads 就是互相重复了,因为他们序列完全相同,这里说明一下,理论上 A 和 B 片段虽然两端被测出来的序列完全相同,中间没有被测到的碱基我们并不知道其序列是否也一样,可能相同也可能不同,我们不得而知,但是现在我们只拿到了文库片段 A/B 两端的序列,所以只能根据现有的信息判断 A/B 就是重复的,这也是 NGS 测序读长短的弊端之一。片段 C 虽然其中一向序列与 A B 重复,但是 C 片段文库片段比 A/B 长,另外一向的序列与 A/B 不同,因此不算 duplicate。
为什么会有 duplicate
要弄清楚这个问题,需要从 NGS 数据产出流程说起:
- 基因组核酸提取
- 基因组 DNA 随机打断,最常用的是超声打断。
- 被打断的 DNA 片段经历末端修复,3' 加A,两端加接头,选择特定大小片段文库进行 PCR 扩增(通过 PCR 扩增选择性提高加上了接头的文库分子数量)。
- 文库上机与 flowcell 上引物结合,经历桥式 PCR 扩增形成 cluster 。
- 进行 SBS 测序,光学信号捕获,生成序列。
我们首先假设基因组核酸提取是完整的基因组,打断是完全随机的(通常是这样的)。
在第 3 步,PCR 扩增时同一个文库分子会产生多个相同的拷贝,这是 duplicate 的主要来源(PCR duplicate)。
第 4 步,文库中 DNA 片段与 flowcell 上引物结合,来源于同一个 DNA 片段的多个拷贝都结合到 flowcell 上,这样会导致生成多个相同的 cluster,测序时也就有多个相同的序列被测出来,这些相同的序列就是 duplicate。
同在第 4 步,生成 cluster 时候一个 cluster 中的 DNA 链可能搭到旁边另外一个 cluster 生成位点上,又长成一个相同的 cluster ,这也是 duplicate 的一个来源(Hiseq4000之后的 flowcell 会有的 cluster duplicate)。
第 5 步,一个 cluster 测序时的捕获的荧光亮点由于形状奇特,可能被软件当成两个荧光点来处理,这也产生了两条完全相同的 reads。这个过程中可能产生完全相同的 reads。(光学 duplicate)
由此我们知道,PCR duplicate 特点是随机分布于 flowcell 表面,光学 duplicate 特点是它们都来自 flowcell 上位置相邻的 cluster 。cluster 的位置被记录在 Fastq 文件 @seq-id 这一行中。
下图的右下角还有一种 duplicate 来源,sister? 这种一个文库分子的两条互补链同时都与 flowcell 上的引物结合分别形成了各自的 cluster,最后产生的两对 reads 完全反向互补,map 到参考基因组也分别在正负链上的相同位置,有的分析中也算 duplicate,虽然我遇到的这种正负链测序结果通常是不算 duplicate 的。

另外,据说 NextSeq 平台上出现过由于荧光信号捕获相机移动位置不够,导致 tile 边缘被重复拍摄,每次采样区域的边缘由于重复采样而出现的 duplicate,下图中蓝色点代表 duplicate ,在 tile 两侧明显富集。Illumina 公司回应说这没毛病,符合预期……

总结一下,duplicate 的产生即有可能来自实验过程,也有可能来自测序仪。
PCR 将模板扩增了数千倍,但数据中 duplication 率只有 15%
我曾经有这样的疑惑,为什么文库构建过程中的 PCR 将每个文库分子都扩增了上千倍,以 PCR 10个循环为例 2^10= 1024 ,但是实际测序数据中 duplication 率并不高(低于20%)。后来我看到一篇文章从统计概率的角度详细探讨了一下 duplication 率的影响因素,顺便一提,这个博主的故事也很令人佩服。
PCR 的过程中不同长度的文库分子被扩增的效率不同(GC 太高或 AT 含量太高都会影响扩增效率),PCR 更倾向于扩增短片段的文库分子,这里先不考虑文库片段扩增效率的差异,把问题简化一下,假设所有文库分子扩增效率都相同。PCR duplicate 的主要来源是同一个文库分子的不同拷贝都在 flowcell 上生成了可以被测序的 cluster ,导致同一个分子的序列被测序仪读取多次。那么为何在每个分子都有上千个拷贝的情况下,实际却很少出现同一分子的多个拷贝被测序的情况呢?主要原因就是文库中 unique 分子的数量比被 flowcell 上引物捕获的分子数量多很多,直白点说就是 flowcell 上用于捕获文库分子的引物数量太少了,两者不在同一个数量级,导致很少出现同一个文库分子的多个拷贝被 flowcell 上引物捕获生成 cluster。
假设文库中所有分子与引物的结合都是随机的,简化一下就相当于,一个箱子中有 n 种颜色的球(文库中的 n 种 unique 分子),每种颜色有 1000 个(PCR 扩增的,随 cycle 数变化),从这个箱子中随机拿出来 k 个球(最终测序得到 k 条 reads),其中出现相同颜色的球就是 duplicate,那么 duplication 率就可以根据有多少种颜色的球被取出 0,1,2,3…… 次的概率计算,可以近似用泊松分布模型来描述。
以人全基因组重测序 30X 为例,PE150 需要约 3x10^8条 reads ,文库中 unique 分子数其实可以通过上机文库的浓度和体积(外加 PCR 循环数)计算出来,这里用近似值 3.5x10^10 个 unique 分子。每个 unique 分子期望被测序的次数是 3x108/3.5x1010 = 0.0085 ,每个 unique 分子被测 0,1,2,3… 次的概率如下图:
> x <- seq(0,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)

由于 unique 分子数量太多,被测 0 次的概率远高于 1 和 2 次,我们去除 0 次的看一下:
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.0085),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)
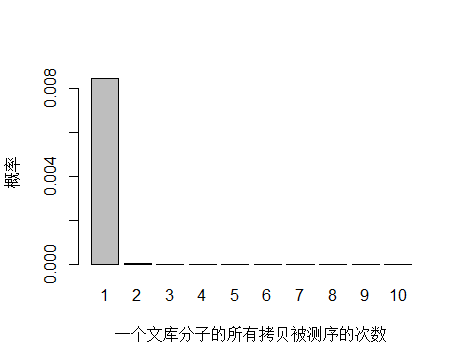
unique 分子被测序 1 次的概率远大于 2次及以上,即便一个 unique 分子被测序 2 次,我们去除 duplicate 时候还会保留其中一条 reads。
如果降低文库中 unique 分子数量到 4.5x10^9 个,PCR 循环数增加以便浓度达到跟上面模拟的情况相同,测序 reads 数还是 3x10^8 条,每个 unique 分子预期被测序的次数是 3x108/4.5x109 = 0.067 。
> x <- seq(1,10,1)
> xnames <- as.character(x)
> xlab <- "一个文库分子的所有拷贝被测序的次数"
> ylab <- "概率"
> barplot(dpois(x,lambda = 0.067),
+ names.arg = xnames,
+ xlab = xlab,
+ ylab = ylab)

unique 分子数量减少,被测序 2次的概率增大,duplication 率显然也会增高。
到这里已经可以很明白的看出 duplication 率主要与文库中 unique 分子数量有关,所以建库过程中最大化 unique 分子数是降低 duplication 率的关键。文库中 unique 分子数越多,说明建库起始量越高,需要 PCR 的循环数越少,而文库中 unique 分子数越少,说明建库起始量越低,需要 PCR 的循环数越多,因此提高建库起始量是关键。