一、前言
前段时间主管让我在GEO数据库弄个表达图谱,准备接下来做各种转录组分析,现在回过头来,觉得这个东西比较简单,十分适合上手练习。思考了一下,决定写下来分享给大家。
注意了,我所说的都是实际工作中会用到的,大家好好看好好学,不过领导有要求不能完全写工作内容,所以我会有修改。
二、什么是GEO数据库
GEO数据库全称GENE EXPRESSION OMNIBUS,是由美国国立生物技术信息中心NCBI创建并维护的基因表达数据库。它创建于2000年,收录了世界各国研究机构提交的高通量基因表达数据,也就是说只要是目前已经发表的论文,论文中涉及到的基因表达检测的数据都可以通过这个数据库中找到。
我是利用NCBI中GEO DataSets查找GEO数据库资料,举个很简单的例子,我想找胃癌相关的疾病资料、研究文献,那么你可以直接搜索gastric carcinoma,当然如果这是一种疾病的话,那你可以直接搜索这种疾病的简称。
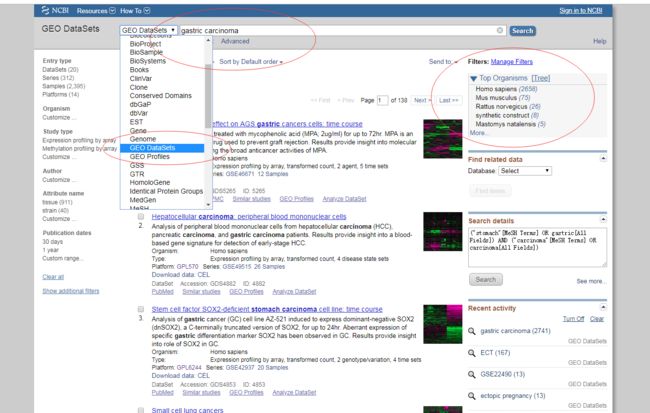
如果你只想关注人相关的研究,请在右方选择类型。
到这里,你必须了解一些比较重要的东西,比如
GEO上有四类数据GSM, GSE, GDS, GPL
1.GSM是单个样本的实验数据
2.GDS是人工整理好的关于某个话题的GSM的集合,一个GDS中的GSM的平台是一样的
3.GSE是一个实验项目中的多个芯片实验,可能使用多个平台
4.GPL是芯片的平台,如Affymetrix, Aglent等
我说简单点,一般我们会利用GSE号在(点击上图GES会直接跳转到GEO数据库)GEO数据库中查找资料,那么GSE号就是我们记录一个实验项目(就是NCBI每一篇文章啦)的编号,如果这个文献有多个芯片平台实验,那么就会产生多个GPL,GPL就是对应你所使用的芯片平台信息。GDS用得比较少,稍微理解一下就行。
三、数据汇总
这部分跟我的工作有关,只是想了解GEO数据挖掘使用方法可以跳过不看。
如果你找的数据是一个比较热门或者比较多人研究的话,你就会得到几百个或者上千个文献,如果我们要对这些文献进行汇总整理,这样做的工程量非常大。你可能需要的东西:GPL、GSE、样本量、样本类型、题目、样本处理方法,甚至文章得出什么结论。这里我用了我比较擅长的爬虫来完成这个任务,对于大批量资料汇总,能活用自己的技能真的太好不过了,我会另起一篇文章单独讲怎么爬NCBI的数据。
四、GEO数据下载
为了完成GEO数据挖掘,做基因表达谱构建,我们必须要下两个文件(如图)
1.GPL探针文件
2.表达矩阵(Series Matrix File)

有萌新可能会说,表达矩阵人家不是做好了吗,我还做啥?对的,这是别人已经处理过的表达矩阵,我们只需要再添加上Symbol就可以完成最简单的GEO数据挖掘——基因表达图谱了。
是不是很简单?
对,就是这么简单,你可以关掉不看了。
我跟你讲,如果你运气好,得到的GPL的文件上就会有对应的Symbol,如果没有,哈,那就八仙过海各显神通吧!
五、数据分析(代码块)
鸽了这么久居然还有人看这篇文章,那好,我就把遇到的坑都和大家讲一下
我做数据分析遇到了很多平台,其他他们的处理方法都不大一样,我一一介绍吧
以GPL570为例,这个平台的数据最多,也是最规整的。
首先我们读取GPL文件和GSE文件
setwd("工作路径")
Sys.setlocale('LC_ALL','C')
#读取GPL文件
GPL_table = read.table('GPL文件地址',sep = "\t",
comment.char = "#", stringsAsFactors = F,
header = T, fill = TRUE, quote = "")
第一步,你们先登录到原网页中看看下载的GPL文件大小对不对,因为NCBI外网连接可能存在网络中断的情况,文件具体大小可以再这里看到,比如说54675行、合计7.7582M等
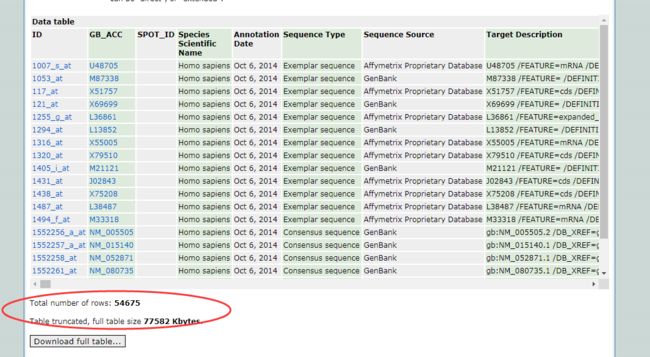
顺便提一下,下载GPL是下面那个Download full table...按钮,如果不提供直接下载,而是加载到网页上的平台,你们直接CTRL+A复制到txt里,一样的,不过注意一定要等NCBI加载完。GSE同理,下载完数据记得都要核查数据大小。
第二步,核查GPL探针文件信息。为了完成基因表达图谱,就一定要看探针文件是否有Gene Symbol的信息,我特地拿570来说,不仅是这个平台的资料最多,而且信息也很完整,因为有一些平台只有GB_ACC,网上有方法回对GB_ACC到基因名称,这里就点一下。

第三步,准备合并表达矩阵。细分下来的步骤就是在GPL中找出基因列和ID列。merge到GSE中,替换掉原来的探针列。这个是我做好的代码,大家改一改自己的路径就可以用了。
setwd("路径")
Sys.setlocale('LC_ALL','C')
#读取GPL文件
GPL_table = read.table('GPL570.txt',sep = "\t",
comment.char = "#", stringsAsFactors = F,
header = T, fill = TRUE, quote = "")
#读取GSE文件
GSE4100 <- read.table('GSE4100_series_matrix.txt',sep = "\t",
comment.char = "!", stringsAsFactors = F,header = T, fill=TRUE)#43931
#表达矩阵制作(已有完整基因名称)
ID_Sybmol = GPL_table[,c(1,11)] #GPL对应ID列
colnames(ID_Sybmol)[2]="Symbol" #更改名称为Symbol,主要是为了对其下面的求平均函数
#合并ID与基因列
Exp = merge(ID_Sybmol,GSE4100,by.x = "ID",by.y = "ID_REF",all=T) #45782个
Exp = Exp[,-1]
到这里,就到表达矩阵最核心的地方,数据居然有4万多行,但基因实际上只有2万多个,这是咋回事?
其实你看一下数据就会发现,很多基因是重复的,而且有些是一对多的探针。
1.所谓一对多,就是一个探针文件对应N个基因,这种探针我们的方法是直接去除。
2.基因重复,我们会以求平均的方式去除过多的行。
3.空值,0值,我都会去除。
4.数据归一化(Normalization),目的是为了后去的去批次差异(batch effect),如果你不懂,那请你百度。
因此第四步,数据清洗,我上完整的数据清洗代码
#数据过滤
Exp = Exp[Exp$Symbol != "",] #45782
Exp = na.omit(Exp) #45782
#去除
Exp1 = data.frame(Exp[-grep("/",Exp$"Symbol"),]) #去一对多,grep是包含的意思,-就是不包含,symbol列不包含 #42984
#求平均值
meanfun <- function(x) {
x1 <- data.frame(unique(x[,1]))
colnames(x1) <- c("Symbol")
for (i in 2:ncol(x)){
x2 <- data.frame(tapply(x[,i],x[,1],mean))
x2[,2] <- rownames(x2)
colnames(x2) <- c(colnames(x)[i], "Symbol")
x1 <- merge(x1,x2,by.x = "Symbol",by.y = "Symbol",all=FALSE)
}
return(x1)
}
Exp2<- meanfun(Exp1)#23520
# 查看数据
par(cex = 0.7)
n.sample=ncol(Exp2[,-1])
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
boxplot(Exp2[,-1], col = cols,main="expression value",las=2)
write.table(Exp2,"Exp_Original.txt",row.names = F,quote = F,sep="\t")
# 校正
row.names(Exp2) = Exp2[,1]
Exp2 = log(Exp2[,-1]) # 我使用的是Log、这里面大家用的会是log2,目的一样,但是最后出的数据集中度不一样,大家一定要注意
# 测试校正数据
par(cex = 0.7)
n.sample=ncol(Exp2)
if(n.sample>40) par(cex = 0.5)
cols <- rainbow(n.sample*1.2)
boxplot(Exp2, col = cols,main="expression value",las=2)
Symbol = row.names(Exp2)
Exp_test = cbind(Symbol,Exp2)
write.table(Exp_test,"Exp.txt",row.names = F,quote = F,sep="\t")
完了我们的表达矩阵就大功告成啦一共23520个。