仅针对人类而言。
虽然一直对GO和KEGG不感冒,但流程化的分析还是要做的。主要包括两部分:
- 将基因编号转为ENTREZID(具有唯一性):基因编号来自ANNOVAR的注释结果,建议别用SYMBOL,因为这种名称特异性较差,在转成ENTREZID时可能出现不唯一的现象。symbol与entrezID并不是绝对的一一对应的
- 利用ClusterProfiler进行富集分析:Y叔更新快,不用担心数据库过时,操作方便,出图好看易调节。
各类数据库的ID,非常多,常见(用)的基本都包含在library(org.Hs.eg.db)中了。
![这么多种ID][1]
01 基因编号转换
人类的数据可以利用library(org.Hs.eg.db)来进行转换。另外,org.db上有20个物种的数据库可供使用
http://bioconductor.org/packages/release/BiocViews.html#___OrgDb
***强烈建议看下官方文档,针对数据库的使用有非常详细的介绍,好用~
将ENSEMBL编号转换为ENTREZID
输入文件example_ensGene:ENSEMBL的列表
source("http://bioconductor.org/biocLite.R")
biocLite(org.Hs.eg.db) #此为人类基因编号系统;mouse为org.Mm.eg.db
library(org.Hs.eg.db)
keytypes(org.Hs.eg.db) #查看基因编号系统名称
EG2Ensembl=toTable(org.Hs.egENSEMBL) #将ENTREZID和ENSEMBL对应的数据存入该变量
#gene_id ensembl_id
#1 ENSG00000121410
ens=read.table("example_ensGene")
#ENSG00000092621
ens=ens$V1
geneLists=data.frame(ensembl_id=ens)
results=merge(geneLists,EG2Ensembl,by='ensembl_id',all.x=T)
id=na.omit(results$gene_id) #提取出非NA的ENTREZID
gene=id
#[1] "728642" "728642" "728642" "728642" "5725" "23357"
将SYMBOL转为ENTREZID
自定义函数,然后批量转换,来自生信技能树中JIMMY的贴子,亲测可用。
geneIDannotation <- function(geneLists=c(1,2,9),name=T,map=T,ensemble=F,accnum=F){
## input ID type : So far I just accept entrezID or symbol
## default, we will annotate the entrezID and symbol, chromosone location and gene name
suppressMessages(library("org.Hs.eg.db"))
all_EG=mappedkeys(org.Hs.egSYMBOL)
EG2Symbol=toTable(org.Hs.egSYMBOL)
if( all(! geneLists %in% all_EG) ){
inputType='symbol'
geneLists=data.frame(symbol=geneLists)
results=merge(geneLists,EG2Symbol,by='symbol',all.x=T)
}else{
inputType='entrezID'
geneLists=data.frame(gene_id=geneLists)
results=merge(geneLists,EG2Symbol,by='gene_id',all.x=T)
}
if ( name ){
EG2name=toTable(org.Hs.egGENENAME)
results=merge(results,EG2name,by='gene_id',all.x=T)
}
if(map){
EG2MAP=toTable(org.Hs.egMAP)
results=merge(results,EG2MAP,by='gene_id',all.x=T)
}
if(ensemble){
EG2ENSEMBL=toTable(org.Hs.egENSEMBL)
results=merge(results,EG2ENSEMBL,by='gene_id',all.x=T)
}
if(accnum){
EG2accnum=toTable(org.Hs.egREFSEQ)
results=merge(results,EG2MAP,by='gene_id',all.x=T)
}
return(results)
}
geneIDannotation()
geneIDannotation(c('TP53','BRCA1','KRAS','PTEN'))
一个超级方便快捷的ID转换方法
利用ClusterProfiler中的bitr
require(ClusterProfiler)
x <- c("GPX3", "GLRX", "LBP", "CRYAB", "DEFB1", "HCLS1", "SOD2", "HSPA2",
"ORM1", "IGFBP1", "PTHLH", "GPC3", "IGFBP3","TOB1", "MITF", "NDRG1",
"NR1H4", "FGFR3", "PVR", "IL6", "PTPRM", "ERBB2", "NID2", "LAMB1",
"COMP", "PLS3", "MCAM", "SPP1", "LAMC1", "COL4A2", "COL4A1", "MYOC",
"ANXA4", "TFPI2", "CST6", "SLPI", "TIMP2", "CPM", "GGT1", "NNMT",
"MAL", "EEF1A2", "HGD", "TCN2", "CDA", "PCCA", "CRYM", "PDXK",
"STC1", "WARS", "HMOX1", "FXYD2", "RBP4", "SLC6A12", "KDELR3", "ITM2B")
eg = bitr(x, fromType="SYMBOL", toType="ENTREZID", OrgDb="org.Hs.eg.db")
head(eg)
02 GO富集分析
over-representation test:enrichGO()
library(clusterProfiler)
ALL <- enrichGO(gene, "org.Hs.eg.db", keyType = "ENTREZID",ont = 'ALL',pvalueCutoff = 0.05,pAdjustMethod = "BH", qvalueCutoff = 0.1, readable=T) #一步到位
BP<-enrichGO(gene, "org.Hs.eg.db", keyType = "ENTREZID",ont = "BP",pvalueCutoff = 0.05,pAdjustMethod = "BH",qvalueCutoff = 0.1, readable=T) #3种分开进行富集
MF <- enrichGO(gene, "org.Hs.eg.db", keyType = "ENTREZID",ont = "MF",pvalueCutoff = 0.05,pAdjustMethod = "BH",qvalueCutoff = 0.1, readable=T)
CC <- enrichGO(gene, "org.Hs.eg.db", keyType = "ENTREZID",ont = "CC",pvalueCutoff = 0.05,pAdjustMethod = "BH",qvalueCutoff = 0.1, readable=T)
#gene: a vector of entrez gene id
#"org.Hs.eg.db":OrgDb
#keyType:keytype of input gene
#ont: One of "MF", "BP", and "CC" subontologies.
#pvalueCutoff:Cutoff value of pvalue.
#pAdjustMethod: one of "holm", "hochberg", "hommel", "bonferroni", "BH", "BY", "fdr", "none"
#universe: background genes
#qvalueCutoff: qvalue cutoff
#minGSSize: minimal size of genes annotated by Ontology term for testing.
#maxGSSize: maximal size of genes annotated for testing
#readable: whether mapping gene ID to gene Name(Symbol)
#pool: If ont=’ALL’, whether pool 3 GO sub-ontologies
write.table(as.data.frame(ALL@result), file="GOALL.txt",quote=FALSE)
#可以对富集结果进行去冗余,方便查看关键信息;无法针对ALL进行去冗余(我也不晓得为啥)
CC_simp <- simplify(CC, cutoff=0.7,by="p.adjust",select_fun=min)
BP_simp <- simplify(BP, cutoff=0.7,by="p.adjust",select_fun=min)
MF_simp <- simplify(MF, cutoff=0.7,by="p.adjust",select_fun=min)
write.table(as.data.frame(CC_simp@result), file="GO_simp.txt")
write.table(as.data.frame(BP_simp@result), file="GO_simp.txt",append=T,col.names=F)
write.table(as.data.frame(MF_simp@result), file="GO_simp.txt",append=T,col.names=F)
做出好看的泡泡图
p=dotplot(ALL,x="count",
showCategory = 14,colorBy="pvalue") #showCategory即展示几个分类,最好都展示(取ALL@result的行数)
library(ggplot2)
library(Cairo)
CairoPDF("enrichGO.pdf",width=10,height=8) #PDF格式非常棒,可在PS中调整dpi
p=dotplot(ALL,showCategory = 14,
colorBy="pvalue",
font.size=18)
p + scale_size(range=c(2, 8)) #设置点的大小
#showCategory即展示几个分类,最好都展示(取ALL@result的行数)
#font.size设置文字大小
dev.off()
![enrichGO.png-182.3kB][2]
#条形图
barplot(ALL, showCategory=15,title="EnrichmentGO_ALL")#条状图,按p从小到大排的
(这个图...好丑)
![Rplot.jpeg-143kB][3]
03 KEGG富集
kk <- enrichKEGG(gene = gene,
organism ='hsa',
pvalueCutoff = 0.05,
qvalueCutoff = 0.1,
minGSSize = 1,
#readable = TRUE ,
use_internal_data =FALSE)
write.table(as.data.frame(kk@result), file="test_kk.txt")
p=dotplot(kk,showCategory = 14,colorBy="pvalue",font.size=18)
效果同上图
04 Reactome pathway enrichment analysis
#install
source("https://bioconductor.org/biocLite.R")
biocLite("ReactomePA")
require(org.Mm.eg.db)
keytypes(org.Mm.eg.db)
EG2Ensembl=toTable(org.Mm.egENSEMBL)
#convert gene ID
query=read.table("geneList.txt")
#ENSMUSG00000043572
#ENSMUSG00000040035
#ENSMUSG00000019564
#ENSMUSG00000042446
query=query$V1
geneLists=data.frame(ensembl_id=query)
results=merge(geneLists,EG2Ensembl,by='ensembl_id',all.x=T)
id=na.omit(results$gene_id) #提取出非NA的ENTREZID
gene=id
#Pathway enrichment analysis
require(ReactomePA)
x <- enrichPathway(gene=gene,pvalueCutoff=0.05, readable=T,organism = "mouse")
head(as.data.frame(x))
# ID Description GeneRatio BgRatio pvalue p.adjust qvalue
#R-MMU-422475 R-MMU-422475 Axon guidance 12/74 278/9315 1.658138e-06 0.0005073902 0.0004415883
#R-MMU-163685 R-MMU-163685 Integration of energy metabolism 7/74 90/9315 6.607488e-06 0.0010109457 0.0008798392
write.table(x,file="ReactomePA.xls",quote=F,sep="\t")
#visulization
barplot(x,showCategory=24)
dotplot(x,showCategory=24)
emapplot(x) #enrichment map
cnetplot(x, categorySize="pvalue", foldChange=gene) #cnetplot(x, categorySize="pvalue", foldChange=geneList)
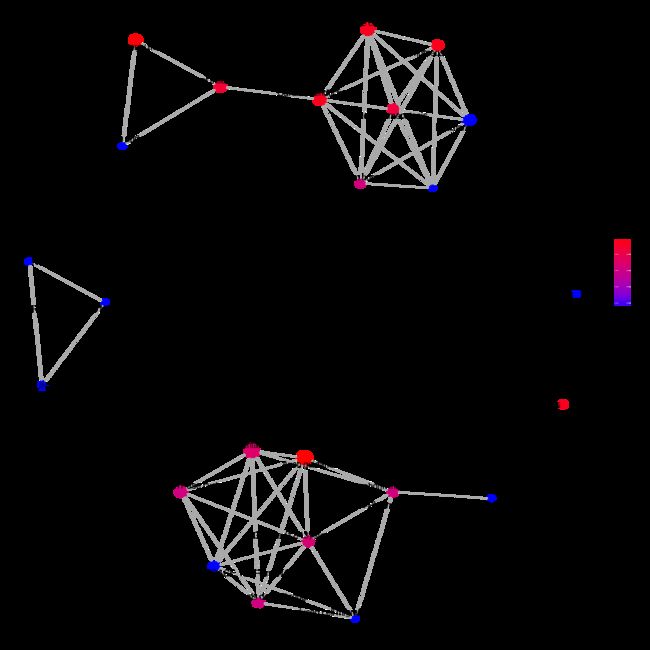
ReactomePA_map.png
Note: 这两个包也可以用来做GSEA
[1]: http://static.zybuluo.com/fatlady/panpe77ea0yw9iqmgt0vt768/image_1casoc2uevrp6gr1f5b88mrqr9.png
[2]: http://static.zybuluo.com/fatlady/lmu9a8kk8bm1bm3psomi4hpu/rare_enrichGO.png
[3]: http://static.zybuluo.com/fatlady/x4btt326zwh3ay0tcj4bk5y3/Rplot.jpeg